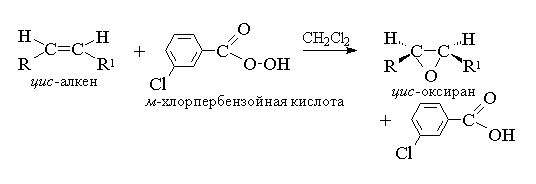
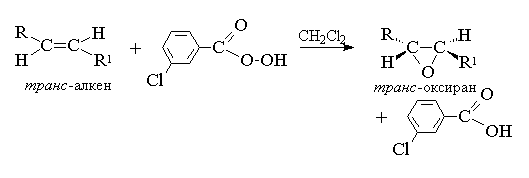
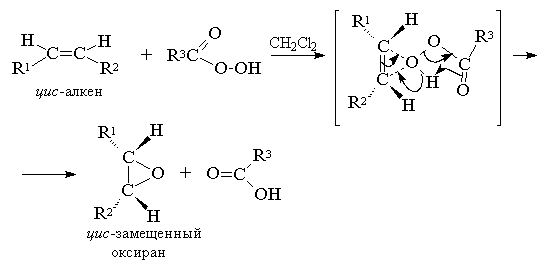
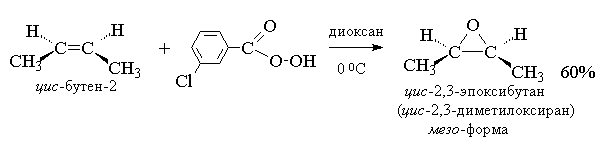
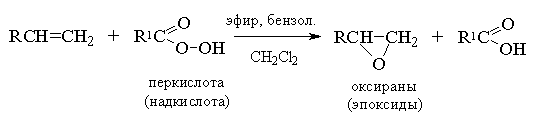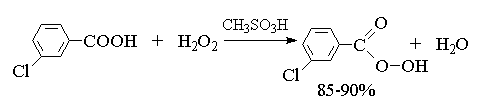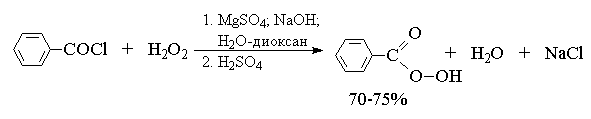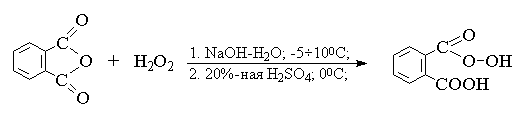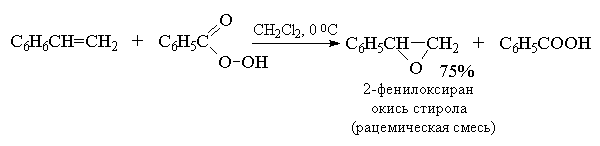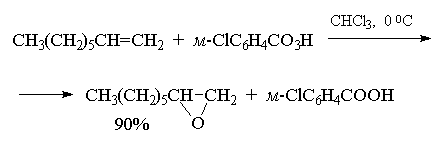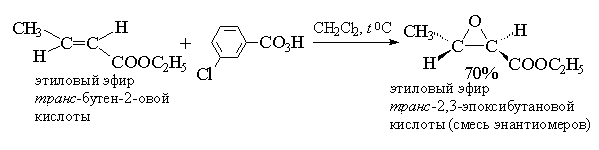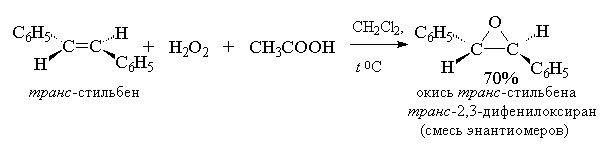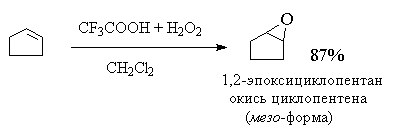2. анти-Гидроксилирование
Трехчленное кольцо оксиранов легко
раскрывается под действием самых разнообразных
нуклеофильных реагентов. Эти реакции подробно
будут обсуждаться в разделе, посвященном
ациклическим и циклическим простым эфирам. Здесь
же будет рассматриваться только гидролиз
оксиранов. Гидролиз оксиранов катализируется
как кислотами, так и основаниями. В обоих случаях
образуются вицинальные диолы, т. е. гликоли.
При кислотном катализе в первой стадии
происходит протонирование атома кислорода
оксирана с образованием циклического
оксониевого катиона, который раскрывается в
результате нуклеофильной атаки молекулы воды:
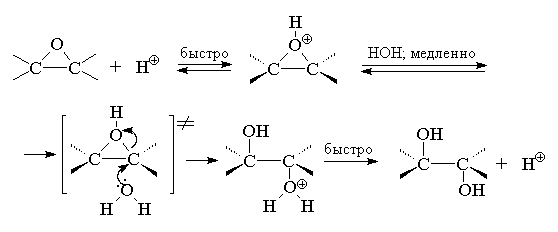
Ключевой стадией в раскрытии кольца,
определяющей скорость всего процесса, является
нуклеофильная атака водой на протонированную
форму оксирана. С точки зрения механизма этот
процесс аналогичен раскрытию бромониевого иона
при нуклеофильной атаке бромид-иона или другого
нуклеофильного агента. С этих позиций
стереохимическим результатом должно быть
образование транс-гликолей при расщеплении
циклических эпоксидов. Действительно, при
кислотно-катализируемом гидролизе
циклогексеноксида или циклопентеноксида
образуются исключительно транс-1,2-диолы.
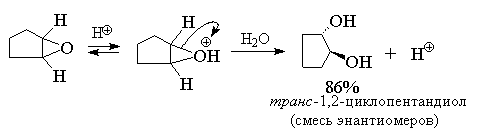
Таким образом, двухстадийный процесс
эпоксидирования алкена с последующим кислотным
гидролизом эпоксида суммарно соответствует
реакции анти-гидроксилирования алкенов.
Обе стадии анти-гидроксилирования алкенов
можно совместить, если алкен обрабатывать водной
30-70%-ной перекисью водорода в муравьиной или
трифторуксусной кислоте. Обе эти кислоты
являются достаточно сильными для того, чтобы
вызвать раскрытие оксиранового цикла.
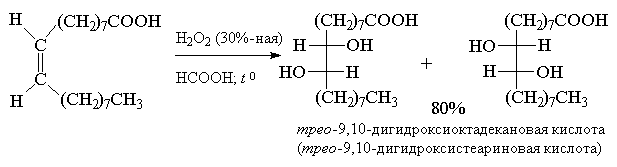
Раскрытие оксиранового кольца, катализируемое
основанием, также приводит к образованию
циклических транс-гликолей.
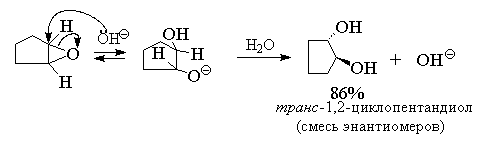
Следовательно, двухстадийный процесс
эпоксидирования алкенов с последующим щелочным
гидролизом эпоксидов также является реакцией анти-гидроксилирования
алкенов.
3. син-Гидроксилирование
Некоторые соли и оксиды переходных металлов в
высших степенях окисления являются эффективными
реагентами син-гидроксилирования двойной
связи алкена, когда обе гидроксильные группы
присоединяются с одной и той же стороны двойной
связи. Окисление алкенов перманганатом калия -
один из старейших методов син-гидроксилирования
двойной связи - продолжает широко
использоваться, несмотря на свойственные ему
ограничения. Цис-1,2-циклогександиол был
впервые получен В.В. Марковниковым в 1878 году
гидроксилированием циклогексена водным
раствором перманганата калия при 0 0С.
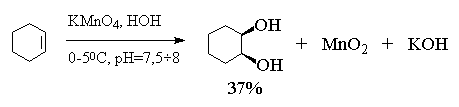
Этот метод в дальнейшем получил развитие в
работах русского ученого Е.Е. Вагнера, поэтому син-гидроксилирование
алкенов под действием водного раствора
перманганата калия носит название реакции
Вагнера. Перманганат калия является сильным
окислителем, способным не только
гидроксилировать двойную связь, но и расщеплять
образующийся вицинальный диол. Для того, чтобы по
возможности избежать дальнейшего расщепления
гликолей, необходимо тщательно контролировать
условия реакции. Выходы гликолей при этом обычно
невелики (30-60%). Наилучшие результаты достигаются
при гидроксилировании алкенов в слабощелочной
среде (рН~8 9) при 0-5 0С
разбавленным 1%-ным водным раствором KMnO4.
9) при 0-5 0С
разбавленным 1%-ным водным раствором KMnO4.
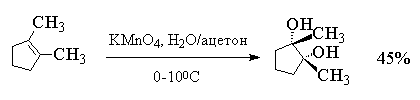
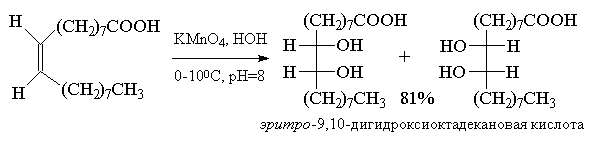
Первоначально при окислении алкенов
перманганатом калия образуется циклический эфир
марганцевой кислоты, который немедленно
гидролизуется до вицинального диола.
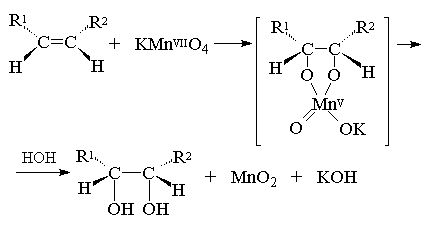
Циклический эфир марганцевой кислоты как
интермедиат не был выделен, однако его
образование следует из экспериментов с меченым 18О
перманганатом калия: оба атома кислорода в
гликоле оказываются мечеными при окислении
алкена KMn18O4. Это означает, что оба
атома кислорода переходят от окислителя, а не из
растворителя - воды, что находится в хорошем
соответствии с предлагаемым механизмом.
Другой метод син-гидроксилирования алкенов
под действием оксида осмия (VIII) OsO4 был
предложен Р. Криге в 1936 году. Тетраоксид осмия
представляет собой бесцветное, летучее,
кристаллическое вещество, хорошо растворимое в
эфире, диоксане, пиридине и др. органических
растворителях. При взаимодействии тетраоксида
осмия с алкенами в эфире или диоксане образуется
черный осадок циклического эфира осмиевой
кислоты - осмат, который легко может быть
изолирован в индивидуальном виде. Присоединение
OsO4 к двойной связи заметно ускоряется в
растворе в пиридине. Разложение осматов до
вицинальных гликолей достигается действием
водного раствора гидросульфита натрия или
сероводородом.
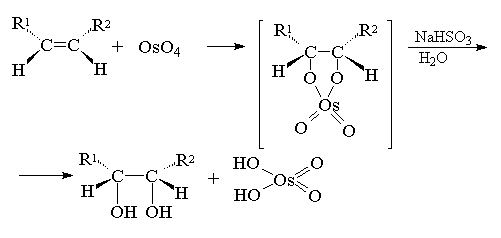
Выходы продуктов син-гидроксилирования
алкенов в этом методе значительно выше, чем при
использовании перманганата в качестве
окислителя. Важным достоинством метода Криге
является отсутствие продуктов окислительного
расщепления алкенов, характерного для
перманганатного окисления.
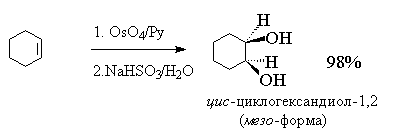
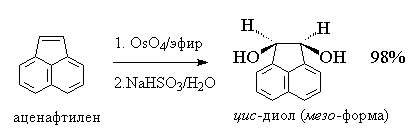
Тетраоксид осмия очень дорогой и
труднодоступный реагент, к тому же он токсичен.
Поэтому оксид осмия (VIII) используется при синтезе
малых количеств трудно доступных веществ с целью
получения наиболее высокого выхода диола. С
целью упрощения син-гидроксилирования
алкенов под действием OsO4 была разработана
методика, позволяющая использовать лишь
каталитические количества этого реагента.
Гидроксилирование алкенов осуществляется с
помощью перекиси водорода в присутствии OsO4,
например:
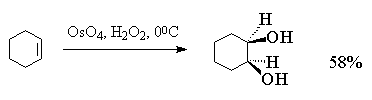
В заключение этого раздела приведем
стереохимические отношения между алкеном цис-
или транс-конфигурации и конфигурацией
образующегося вицинального диола, который может
быть цис- или транс-изомером, эритро-
или трео-формой, мезо- или D,L-формой в
зависимости от заместителей в алкене:

Аналогичные стереохимические отношения
наблюдаются и в других реакциях син- или анти-присоединения
по кратной связи водорода, галогенводородов,
воды, галогенов, гидридов бора и др. реагентов.