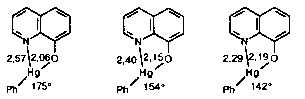
Химическое и физическое
взаимодействие атомов.
Специфические межмолекулярные контакты
В начале XIX века в химии сформировались представления, согласно которым во всякой совокупности близко расположенных (контактирующих) атомов есть химически связанные атомы. Иными словами, имеются пары атомов, между которыми существуют химические связи. Наряду с этим, естественно, есть и пары атомов, химически не связанных. Позже эти представления привели к понятию молекулы, которая мыслится как совокупность последовательно соединенных атомов, как ансамбль химических связей.
Развитие такого взгляда на химические соединения выдвинуло ряд кардинальных проблем.
Какие атомы считать связанными, а какие – нет, и что из этого следует?
Какова природа сил, точнее потенциалов, обусловливающих химические связи?
Что происходит с химическими связями в ходе разнообразных процессов (химических и физических)?
Эги вопросы обсуждаются вот уже почти два столетия, но ни один из них и поныне нельзя считать полностью решенным.
Впервые достаточно полная концепция, от вечающая на первый вопрос, появилась в 50-х – 60-х годах прошлого столетия в виде "классической теории химического строения" органических молекул; ее создателями были Ф. А. Кекуле (1857), А. С. Купер (1858), А. М. Бутлеров (1861). Для неорганических, в частности, координационных соединений аналогичный подход был развит в работах А. Вернера (1893). Эта самая старшая по возрасту и вместе с тем и до сих пор самая важная для химии модель структуры базируется на колоссальном фундаменте опытных данных, на постоянно пополняемых и корректируемых эмпирических представлениях. Разумеется, далеко не всегда эмпирическая система, описанная в многотомных курсах органической и неорганической химии, дает ясную и правильную картину ансамбля химических связей. Но в большинстве случаев она устраивает химиков и даже тогда, когда эти представления явно отказывают, классическая теория химического строения служит хорошим "нулевым приближением", на фоне которого легче создавать более совершенные схемы.
Для последовательного, научно обоснованного ответа на вопрос о том, какие атомы в химическом веществе следует считать связанными, а какие – нет, предлагались весьма различные подходы и критерии. Одна из новейших методик – градиентный анализ распределения электронной плотности – рассмотрена ниже.
И все же попытка решить этот вопрос, исходя из любой, сколь утодно совершенной модели статичного химического объекта, модели равновесного вещества, заведомо не может привести к полноценным результатам. Формировавшаяся в течение полутора веков эмпирическая система воззрений на совокупность химических связей включает в себя не только характеристики равновесного вещества, но и сведения о его поведении при разнообразных внешних воздействи ях (изменение условий, действие реагентов). Именно такой подход нужен химии.
Таким образом, адекватное описание системы химических связей, имеющихся в ве ществе, требует построения и анализа много численных и весьма сложных потенциальных поверхностей, включая исследование путей реакций и друтих процессов, определение высоты потенциальных барьеров, оценку энергий активации и т.д. Однако даже если бы все это было доступно и легко осуществимо, в конечном итоге мы пришли бы к необходимости создания аппарата (языка), позволяющего суммировать имеющуюся информацию и представить ее в наглядном виде. Но скорее всего тут снова получилось бы нечто похожее на классическую систему химического строения с ее структурными формулами (графами); разумеется, при этом многие графы неорганических веществ (в твердом состоянии) оказались бы бесконечными в одном, двух или трех измерениях, что соответствует цепочечным, слоистым и каркасным структурам.
Сказанное побуждает с глубоким уважением и доверием относиться к картине химических связей, трактуемых на основе классической теории химического строения, - это главное богатство химии. Конечно же, классическая теория должна постоянно корректироваться; временами приходится оговаривать существова ние отдельных химических объектов и даже целых классов веществ и процессов, для которых классические представления неприменимы или недостаточны. С другой стороны, имеет огромное значение и дальнейшее развитие фи зической интерпретации химического связывания, и углубленное квантовомеханическое исследование процессов образования и разрыва химических связей. Только не нужно думать, что в обозримом будущем эти более строгие подходы смогут заменить классическую теорию: очень многие закономерности поведения химических веществ, легко описываемые на языке классической теории химического строения (с учетом количественной структурной информации), оказываются безнадежно сложными для последовательной физической трактовки.
Фактически в рамках обсуждения первого из поставленных выше вопросов неизбежно воз никают и второй, и третий вопросы. В конце 20-х годов XX века, когда возникла квантовая механика, а сразу вслед за ней и квантовая хи мия, впервые обозначился удовлетворительный путь интерпретации химического связывания на основе фундаментального закона природы, вы ражаемого уравнением Шредингера; таким способом удалось объяснить образование молекулы Н2 (В. Г. Гайтлер и Ф. Лондон, 1927)[Электронные концепции химических связей Г. Н. Льюиса (1916) и И. Ленгмюра (1919), базирующиеея на модели атома Н. Бора, сыграли огромную роль в развитии химической теории, они важны и поныне, но, как и классическая теория химического строения, эти концепции имели характер постулатов, основанных на эмпиричееких обобщениях].
Однако это был скорее успех физики, а не химии. Физика продемонстрировала свои возможности в еще одной области и открыла для себя новое поле деятельности. Что же касается химии, то для исследования многообразия химических соединений Хi и зависимостей р(Х) использование квантовой механики не давало заметных преимуществ. Расчетно-теоретическая интерпретация химических связей в мало-мальски сложных системах стала возможна лишь в последние годы, причем проблема точности и однозначности прогноза в настоящее время остается актуальной даже для молекул, содержащих 10 - 15 атомов. Что же касается действительно сложных многоатомных систем, с которыми имеет дело современная химия, то здесь квантовохимические методы обычно не в состоянии адекватно отобразить и тем более предсказать картину химического связывания атомов.
Как уже было сказано, сколько-нибудь полное представление о совокупности химических связей в данной системе можно получить только путем анализа ее динамики. И наоборот, динамика химической системы становится понятной лишь при учете присутствующих связей. Важно подчеркнуть, что не только реакции, но и фазовые переходы и друтие. "физические" процессы дают важную информацию о химических связях.
Существенно позже, чем концепция химических связей, возникли представления о том, что во всяком веществе взаимодействуют не только химически связанные, но другие достаточно близко расположенные атомы - в таких случаях говорят о невалентных (или физических) межатомных взаимодействиях. Точнее, отрывочные суждения о слабых взаимодействиях между молекулами и химически несвязанными атомами появлялись в научной литературе XIX и даже XVIII века. Но лишь после исследований Я. Д. Ван-дер-Ваальса, опубликованных в 1873 г., эти взаимодействия стали рассматриваться как фундаментальное свойство атомно молекулярных систем. В настоящее время хорошо известно, что во многих процессах (в том числе иногда и в химических реакциях) ван дер-ваальсовы взаимодействия играют важную, часто решающую роль.
С позиции последовательного квантовомеха нического подхода природа "химических" и "физических" межатомных взаимодействий одна и та же: это электростатическое взаимодействие, но не в кулоновском понимании, подразумевающем взаимодействие точечных зарядов, а в том смысле, который вытекает из уравнения Шредингера, где используется понятие волновой функции. Однако, если рассматривать типичные ковалентные, ионные или металлические связи и типичные невалентные атом атомные взаимодействия (последние ярче всего проявляются в межмолекулярных контактах), то обнаружатся весьма разные по величине энергетические эффекты, весьма различные структурные последствия этих двух типов взаимодействий.
Вместе с тем, между химическими связями и невалентными взаимодействиями нельзя провести сколько-нибудь определенную границу. Это стало особенно ясно в последние годы, когда появились многочисленные исследования, посвященные "вторичным (дополнительным) химическим связям" (см., например, [10, 11]), с одной стороны, и "специфическим межмолекулярным взаимодействиям" [12], с другой. Ниже приведены примеры этих промежуточных взаимодействий, более сильных, чем ван-дер ваальсовы, но уступающих по своей стабильности обычным ковалентным связям.
Нередко в координационное окружение атома М (чаще всего это атом переходного метал ла), кроме его ближайших соседей Х, образующих с ним обычные ковалентные связи М – Х, входят дополнительные атомы, более удаленные, но все же достаточно сильно взаимодей ствующие с атомом М. Так, 8-оксихинолинат фенилртути, который кристаллизуется в виде двух полиморфных модификаций, причем в одной из них сосуществуют две разновидности молекул, оказывается представлен тремя конформерами, их строение установлено с помощью рентгеноструктурного анализа [13]:
(значения межатомных расстояний указаны в А, расстояния Hg – С приблизительно постоянны и близки к 2,0 А). Здесь прежде всего обращает на себя внимание неравноценность связей атома Hg с соседними атомами, причем связи Hg – О и Hg – С естественно считать обычными кова лентными, а связи Hg – N вторичными. Вместе с тем обнаруживается значительная подвижность рассматриваемой системы. Расстояние Hg...N может укорачиваться и, если считать, что такое укорочение соответствует усилению связи, то в третьей разновидности молекулы вторичная связь уже очень близка к ковалентной. Примечательно, что укорочение связи Hg...N сопровождается удлинением связи Нg – О и уменьшением угла С – Hg – О (это можно трактовать, как переход состояния атома Нg от sp к sp2. Этот пример красиво дополняется тем, что, как установлено в той же работе [13], в кристаллах 2-метил-8-оксихинолината фенил ртути реализуется еще одно промежуточное состояние ртути (Hg...N 2,36, Нg – О 2,16А, С – Hg – О 151,8o).
Нередко встречаются также примеры дополнения координации атома М атомами соседних молекул. При этом иногда расстояние М...Х намного (чуть ли не вдвое) превышает расстояние, характерное для ковалентной связи М – Х, и все же расположение молекул ("логика структуры") дает основания предполагать, что атом атомный контакт М...Х играет особую структурообразующую роль, что, конечно же, должно быть сопряжено с его особой энергетической выгодностью. Однако в подобных случаях лучше говорить не о "вторичных связях", а о "специфических межмолекулярных контактах". Бывает и так, что в подобных ситуациях наблюдается специфическое взаимодействие между атомами металла соседних комплексных молекул. Примерами могут служить кристаллы хелатных соединений никеля – салицилалимината Ni и диметилглиоксимата Ni; в первых присутствуют паркетные молекулярные слои, образованные с помощью контактов Ni...О длиной 3,85 А, во вторых – цепи, содержащие контакты Ni...Ni длиной 3,25А (рис. 5).
Наиболее широко распространенный тип специфических межмолекулярных взаимодей ствий – это, конечно, водородные связи (Н - связи). О них написано много, и в настоящей статье нет надобности развивать эту тему. Полезнее указать на некоторые другие менее известные, но очень характерные типы специфических межмолекулярных контактов, а именно ортогональные бензольные контакты и контакты галоген...галоген.
Взаимно перпендикулярное Т-образное расположение плоских бензольных циклов в кри сталлическом бензоле (рис. 6а) – достаточно часто упоминаемый факт. Однако, как правило, остается неизвестным, что таким же способом располагаются соприкасающиеся фенильные радикалы соседних молекул во многих других органических кристаллах (например,в 2 аминофеноле, рис. 6б). Такое расположение очень часто воспроизводится в самых разных органических веществах – значит, оно обладает особой выгодностью.
Контакты галоген...галоген тоже очень часто играют важную структурообразующую роль.
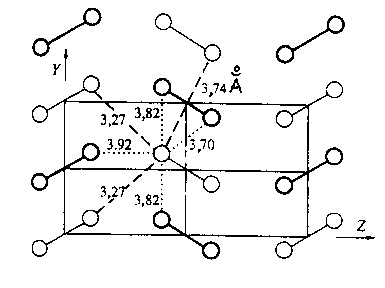
Рис. 8. Кристаллическая структура хлора (проекция па плоскость YZ).
(Молекулы, показанные тонкими линиями, располагаются на высоте х = 0; молекулы, изображенные более толстыми линиями, находятся на высоте х = a/2 (а также на высоте – a/2); приведены кратчайшие межмолекулярные расстояния Cl...Cl при 90 К )
В галогенсодержащих органических
веществах атомы галогенов вовсе не стремятся
рассредоточиться в пространстве, а, напротив,
образуют агрегаты (ленты, стержни, слои), в
которых каждый атом галогена имеет в своем
ближайшем окружении несколько (обычно 4 – 6)
одноименных атомов. Так, в кристаллах  -модификации n-дихлорбензола (рис. 7) молекулы уложены в стопки,
вытянутые вдоль оси Z, стопки упакованы в
слои, параллельные плоскости YZ, а слои
соединяются посредством контактов Cl...C1. Каждый
атом Cl образует четыре таких контакта: один
длиной 3,39 А, два длиной 3,88 А и один длиной 4,17 А.
Ван-дер ваальсов радиус хлора равен 1,90 А [14].
Следовательно, первый контакт существенно
сокращен, а остальные лишь немного превышают
удвоенный ван-дер-ваальсов радиус хлора (2R =3,80 А).
Характерно, что средняя длина указанных четырех
контактов равна 3,83 А; это очень близко к 2R. Совокупность
близко расположенных атомов Cl (Cl-aгpeгaт) в данном
случае представляет собой слой, изображенный на
рис. 7б.
-модификации n-дихлорбензола (рис. 7) молекулы уложены в стопки,
вытянутые вдоль оси Z, стопки упакованы в
слои, параллельные плоскости YZ, а слои
соединяются посредством контактов Cl...C1. Каждый
атом Cl образует четыре таких контакта: один
длиной 3,39 А, два длиной 3,88 А и один длиной 4,17 А.
Ван-дер ваальсов радиус хлора равен 1,90 А [14].
Следовательно, первый контакт существенно
сокращен, а остальные лишь немного превышают
удвоенный ван-дер-ваальсов радиус хлора (2R =3,80 А).
Характерно, что средняя длина указанных четырех
контактов равна 3,83 А; это очень близко к 2R. Совокупность
близко расположенных атомов Cl (Cl-aгpeгaт) в данном
случае представляет собой слой, изображенный на
рис. 7б.
Поскольку специфические контакты галоген...галоген часто встречаются в кристаллах органических галогенпроизводных, они должны реализоваться и в кристаллических галогенах. Действительно, укороченные межмолекулярные расстояния Hal...Hal в кристаллических хлоре, броме и иоде – давно и хорошо известный факт [15]. Система наиболее коротких расстояний Cl...Cl в кристаллическом хлоре показана на рис. 8(см.выше); кратчайший контакт Cl...C1 в этих кристаллах имеет длину 3,27 А, т.е. значительно сокращен по сравнению с 2R(C1). Существование такого короткого контакта (и реализацию орторомбической кристаллической структуры) удается объяснить [16] только вкладом обменного взаимодействия, т.е. частично ковалентным характером этой связи.
Совокупность специфических межмолекулярных контактов в твердых галогенах представляется особенно наглядной при сопоставлении строения изоструктурных кристаллов хлора, брома и иода (рис. 9). Наблюдаемый здесь эффект – постепенное сближение кратчайшего межатомного расстояния d, соответствующего химической связи, и кратчайшего расстояния R между атомами, которые считаются химически несвязанными, – в кристаллохимии называется "уменьшением гетеродесмичности структуры" (от греческого слова "десмос", что значит "связь"). Обращает на себя внимание удивительный и чрезвычайно мало известный факт: естественным продолжением этого структурногo ряда является кристаллический галлий.
Еесли в кристаллической структуре хлора сблизить молекулы, составляющие слой, параллельный плоскости YZ (рис. 8(см.выше) и 9), настолько, что межмолекулярные контакты Hal...Hal станут равны длинам связей Hal – Hal, получится искаженная графитоподобная сетка. Далее, если сблизить слои так, чтобы и межслоевые контакты Hal... Hal по длине стали равны внутрислоевым межатомным расстояниям, то получится структура, очень близкая к наблюдаемой в кристаллах галлия (рис. 10). В этой структуре, разумеется, нет физически реальных молекул и слоев, и термин "слой" при ее описании используется лишь в чисто геометрическом смысле,
Описанную тенденцию – уменьшение гетеродесмичности хорошо передает величина отношения R/d; 1,66 для хлора; 1,46 для брома; 1,29 для иода и 1,09 для галлия. Кристаллический фтор имеет другую структуру, но и он в принципе укладывается в обсуждаемую закономерность в качестве исходного члена ряда.
Кратчайшие межатомные расстояния в кристаллах галогенов и галлия сопоставлены в табл. 4. Как видим, координационное окружение атома в кристаллическом хлоре выражается суммой:
1 (1,98 А) + 2 (3,27 А) + 2 (3,70 А) + 1 (3,74 А) + + 2 (3,82 А) + 2 (3,92 А)
В кристаллическом галлии атом имеет семь приблизительно равноудаленных соседей.
Ясно, что в рассматриваемом ряду веществ закономерно меняется не только пространственное расположение атомов, но и характер химических связей: усиливается обменное взаимодействие атома с его ближайшим окружением. Пользуясь общепринятым делением связей на кова лентные, ионные и м веские [5], можно сказать, что при переходе от флора к увеличивается вклад металлической связи.
Таблица 4. Межатомные расстояния (в А) в кристаллах галогенов и галлия
| Симметр. операция |
 (000) (000) |
b  Z Z |
(0 1/2 0) |
t(1/2 1/2 0) | 21|| Х | a Z |
- |
| Кратность | 1 | 2 | 1 | 4 | 2 | 2 | 2 |
| Иод | 2,72 | 3,50 | 3,97 | 4,24 | 4,34 | 4,41 | - |
| Бром | 2,27 | 3,32 | 3.80 | 4,02 | 3,99 | 4,14 | - |
| Хлор | 1,98 | 3,32 | 3,82 | 3,84 | 3,74 | 3,97 | - |
| Галлий | 2,47 | 2,70 | 2,79 | 2,74 |
Интересно оценить структурообразующую роль каждого из более или менее коротких межатомных контактов в рассмотренных кри сталлах на основе данных о распределении электронной плотности, и в случае хлора мы имеем такую возможность (см. следующий раздел). Однако обстоятельный анализ химическо го связывания в кристаллах со специфическими взаимодействиями пока остается делом будущего.
Итак, чтобы правильно и полно представить весь диапазон межатомных взаимодействий и вытекающее отсюда многообразие структур и свойств химических веществ, необходимо иметь в виду и сильные валентные связи, и слабые невалентные ван-дер-ваальсовы взаимодей ствия, и промежуточные вторичные связи, и специфические межмолекулярные контакты.
Продолжение