|
В.В.Загорский
Трудные темы школьного
курса химии
Элементы химической термодинамики и кинетики
Урок 2 (7)
Второй закон термодинамики
Вполне очевидно, что реакции с суммарным уменьшением энтальпии (экзотермические) могут идти самопроизвольно, как катящийся с горы камень. Однако хорошо известно, что самопроизвольно идут также некоторые реакции, сопровождающиеся увеличением энтальпии и охлаждением реактора (эндотермические).
Для характеристики эндотермических процессов и определения условий их самопроизвольного осуществления была введена новая функция состояния – энтропи'я (от греч. “эн” – “в”, “внутрь” и “тропе” – “поворот”, “превращение”) [ ] [ 1]. Изменение энтропии равно (по определению) минимальной теплоте, подводимой к системе в обратимом (все промежуточные состояния равновесны) изотермическом процессе, деленной на абсолютную температуру процесса: ] [ 1]. Изменение энтропии равно (по определению) минимальной теплоте, подводимой к системе в обратимом (все промежуточные состояния равновесны) изотермическом процессе, деленной на абсолютную температуру процесса:
 S = Q мин. /T S = Q мин. /T
На данном этапе изучения термодинамики следует принять как постулат [ 2], что существует некоторое экстенсивное свойство системы S, называемое энтропи'ей , изменение которого так связано с процессами в системе:
| В самопроизвольном процессе |
S > Q мин. /T |
| В равновесном процессе |
S = Q мин. /T |
| В несамопроизвольном процессе |
S < Q мин. /T |
Для изолированной системы , где dQ = 0, получим:
В самопроизвольном процессе |
S > 0 |
В равновесном процессе |
S = 0 |
В несамопроизвольном процессе |
S < 0 |
В общем случае энтропия изолированной системы или увеличивается, или остается постоянной :
S  0 0
Понятие энтропии возникло из полученных ранее формулировок второго закона (начала) термодинамики. Энтропия – свойство системы в целом, а не отдельной частицы.
Формулировка второго начала по У.Томсону (1851): “в природе невозможен процесс, единственным результатом которого была бы механическая работа, совершенная за счет охлаждения теплового резервуара”.
По Р.Клаузиусу (1850): “ теплота сама по себе не может перейти от более холодного тела к более теплому ” или: “невозможно сконструировать машину, которая, действуя посредством кругового процесса, будет только переносить теплоту с более холодного тела на более теплое”.
Самая ранняя формулировка второго начала термодинамики появилась раньше первого начала, на основании работы французского инженера С.Карно [ ] (1824) и ее математической интерпретации Э.Клапейроном (1834) как КПД (коэффициент полезного действия) идеальной тепловой машины:
КПД = (T 1 - T 2 )/T 1
Карно и Клапейрон [ ] на самом деле сформулировали закон сохранения теплорода – невесомой неуничтожимой жидкости, содержание которой определяет температуру тела. Теория теплорода господствовала в термодинамике до середины XIX века, при этом законы и соотношения, выведенные на основе представлений о теплороде, оказались действительными и в рамках молекулярно-кинетической теории теплоты.
Третий закон термодинамики (постулат Планка)
В 1911 г. Макс Планк [ ] предложил следующий постулат: энтропия правильно сформированного кристалла чистого вещества при абсолютном нуле равна нулю . Этот постулат может быть объяснен статистической термодинамикой , согласно которой энтропия есть мера беспорядочности системы на микроуровне [ ].
Соотношение S = kblnW
часто называют “уравнением Больцмана“, хотя оно выведено М.Планком в 1900 г. [ 3]. В нем W – число различных состояний системы, доступное ей при данных условиях, или термодинамическая вероятность макросостояния системы.
kb = R/NA = 1,38.10-16 эрг/град – постоянная Больцмана
В 1872 г. Л.Больцман [] предложил статистическую формулировку второго закона термодинамики [ ]: изолированная система эволюционирует преимущественно в направлении большей термодинамической вероятности [ 4].
Следует всегда помнить, что второй закон термодинамики не является абсолютным ; он теряет смысл для систем, содержащих малое число частиц, и для систем космического масштаба [ 5]. Второй закон, особенно в статистической формулировке, неприменим к живым объектам , которые представляют собой открытые системы и постоянно уменьшают собственную энтропию, создавая идеально упорядоченные молекулы, например, за счет энергии солнечного света [].
Для живых систем характерна самоорганизация, которую чилийский нейробиолог Умберто Матурана (Humberto Maturana) [] назвал в 1970 г. автопоэз (самосозидание). Живые системы не только сами постоянно удаляются от классического термодинамического равновесия, но и делают неравновесной окружающую среду. Еще в 1965 г. американский специалист по химии атмосферы Джеймс Лавлок (Lovelock) предложил в качестве критерия наличия жизни на Марсе оценивать равновесность состава атмосферы [ 6].
В атмосфере Земли содержатся одновременно кислород (21% по объему), метан (0,00018%), водород (0,00005%), моноксид углерода (0,00001%) – это явно неравновесная смесь при температурах -500 - +400С [ 7] [ ]. Земная атмосфера – открытая система, в формировании которой постоянно участвуют живые организмы.
В атмосфере Марса преобладает углекислый газ (95% - ср. с 0,035% на Земле), кислорода в ней менее 1%, а газы-восстановители (метан) пока не обнаружены [ 8] []. Следовательно, атмосфера Марса практически равновесна – все реакции между содержащимися в ней газами уже осуществились.
Из этих данных Лавлок заключил, что в настоящее время на Марсе жизни нет.
В то же время существует возможность применять законы классической термодинамики к живым объектам. Открытые живые системы на определенных временах можно рассматривать как квазизакрытые по отношению к процессам, для которых быстро устанавливаются локальные равновесия (Гладышев Г.П. [ 9]). Структуры высшей иерархии образуют для более низших “термостат”, в рамках которого процессы можно считать квазиравновесными.
Однако вернемся к “нормальным” химическим системам.
Введение энтропии дало возможность установить критерии, позволяющие определить направление и глубину протекания любого химического процесса (для большого числа частиц в равновесии).
Макроскопические системы достигают равновесия , когда изменение энергии компенсируется энтропийной составляющей:
При постоянном объеме и температуре: Uv = T Sv или (U-TS) F = 0
F – энергия Гельмгольца [ 10] [ ] или изохорно-изотермический потенциал .При постоянном давлении и температуре:
Hp = T S p или (H-TS)  G = 0 G = 0
G энергия Гиббса или свободная энергия Гиббса или изобарно-изотермический потенциал
Изменение энергии Гиббса как критерий возможности химической реакции
Для данной температуры G = H - T S
При G < 0 |
реакция возможна; |
при G > 0 |
реакция невозможна; |
при G = 0 |
система находится в равновесии. |
Возможность самопроизвольной реакции в изолированной системе определяется сочетанием знаков энергетического (энтальпийного) и энтропийного факторов:
Знак H |
Знак S |
Возможность самопроизвольной реакции |
+ |
– |
Нет |
– |
+ |
Да |
– |
– |
Зависит от соотношения H и T S |
+ |
+ |
Зависит от соотношения H и T S |
Имеются обширные табличные данные по стандартным значениям Go и So (для энтропии по третьему закону есть нулевой уровень отсчета и соответственно абсолютные значения), позволяющие вычислить Go реакции.
В случае, если температура отличается от 298 К и концентрации реагентов C – от 1 М, для процесса в общем виде:
aA + bB  xX + yY xX + yY
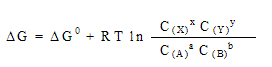
В положении равновесия G = 0 и Go = -RTlnK p , где Kp - константа равновесия
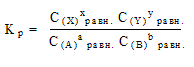
Иногда выражение для константы равновесия называют законом действующих масс (К.М.Гульдберг [ ], П.Вааге [] , 1867 г. ), поскольку в 1867 г. концентрации в работах норвежских ученых назывались “действующими массами”. Применительно к равновесию это не совсем верно [ ] [ (глава 4.7)], поскольку выражение для константы равновесия в большинстве случаев нельзя рассматривать как отношение кинетических уравнений прямой и обратной реакций (см. ниже).
Константа равновесия связана, таким образом, с энергией Гиббса: Kp = exp(- G =o/RT)
Пользуясь приведенными формулами, можно определить температуру, начиная с которой эндотермическая реакция, при которой возрастает энтропия, становится легко осуществимой. Температура определяется из условия:
G o = H o - TSo = 0; T = H o / So
Следует учесть, что для точных расчетов при температурах, заметно отличающихся от 298 К, необходимо использовать для всех термодинамических функций их зависимости от температуры . Например, для реакции:
2 CH4 = C2H4 + 2H2
В первом приближении H o= 48,3 ккал/моль C2H4 ;
во втором приближении H t = 46370 + 6,47T кал/моль [ 11]
“Проблема” слабых электролитов
Диссоциация уксусной кислоты: CH3COOH  CH3COO – + H + CH3COO – + H +
Вещество |
G o298, кДж/моль [ 12] |
CH3COOH |
-377 |
H + |
0 |
CH3COO – |
-369 |
Расчет по закону Гесса: G oреакции = (-369 + 0) - (-377) = + 8 кДж (!)
Но так получится, если все концентрации стандартные – по 1 моль/л
Уточняем концентрацию ионов CH3COO – и H + для одномолярной кислоты.
Константой равновесия в этом случае является константа диссоциации кислоты К дисс. = 1,8*10–5. Тогда :
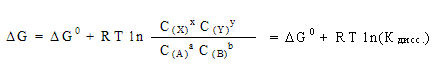
G = -369 + 2,478ln(10–5 ) = -369 + 2,478*(-11,5) = -369 + (-28) = -397
G (реакции) = (-397 + 0) - (-377) = -20 кДж
Это соответствует хорошо известному экспериментальному факту – уксус кислый.
Дополнение 1
Простейшие примеры практически важных термохимических расчетов
Для реакций разложения твердых веществ, сопровождающихся образованием газов (возрастание энтропии), расчет энтальпии может служить критерием предсказания возможности самопроизвольности процесса. такие расчеты важны для оценки опасности некоторых окислителей, применяемых в пиротехнике [ 13] [ ].
Рассмотрим такие расчеты на примере нитрата аммония .
Стандартная энтальпия образования по условному процессу:
| (1) 2H2 + N2 + 1,5 O2 = NH4NO3 |
Ho298= -365 кДж / моль |
| (2) NH4NO3 = N2 + 2 H2O + 0,5O2 |
Ho298= -119 кДж / моль
|
Казалось бы, обратный процесс разложения нитрата аммония эндотермичен. Но при разложении этой соли получается не смесь водорода с кислородом, а вода, образование которой сопровождается выделением энергии. Наиболее вероятна реакция:
Возможны и реакции, ведущие к образованию эндотермичных оксидов азота:
| (3) NH4NO3 = N2O + 2 H2O |
Ho298= -37 кДж / моль |
| (4) NH4NO3 = 0,5 NO2 + 0,75N2 + 2H2O |
Ho298= -102 кДж / моль |
Необходимые табличные данные:
Вещество |
Ho298 , кДж/моль |
H2O (г) |
-242 |
N2O |
+82 |
NO2( г ) |
+34 |
Из приведенных термохимических уравнений видно, что возможен самопроизвольный распад нитрата аммония (аммиачной селитры) с выделением энергии. При попытке нагреть небольшое количество нитрата аммония самопроизвольная реакция не начинается. Однако эта соль начнет разлагаться при нагревании, если к ней добавить катализатор – 5-8% по массе бихромата калия.
Возможно самопроизвольное разложение по другому механизму. Первый широко известный случай такого разложения произошел на складе химического завода в Оппау (Германия, 1921г.). При попытке раздробить слежавшуюся массу смеси нитрата и сульфата аммония небольшими зарядами динамита эта масса взорвалась (сдетонировала), в результате погибло около 600 человек, были разрушены десятки зданий. Вид завода в Оппау после взрыва

С тех пор произошло более 40 описанных в литературе крупных аварий, связанных с детонацией нитрата аммония.
Энтальпия реакции (2) как раз соответствует эмпирическому критерию возможности самопроизвольного устойчивого горения : в реакции должно выделяться около 1,5 кДж на грамм исходных веществ.
Проведем аналогичный расчет для хлората калия:
| (5) KClO3= KCl + 1,5O2 |
 Ho298 =-48 кДж/моль Ho298 =-48 кДж/моль |
Чистый хлорат калия самопроизвольно не разлагается, однако смесь этой соли даже с небольшим количеством горючих веществ (фосфор, сера) чувствительна к трению и удару. Поэтому данная соль сейчас почти не применяется в пиротехнике, за исключением производства спичек.
Когда К.Л.Бертолле [ ] открыл хлорат калия (1786 г.), была предпринята попытка использовать эту соль вместо традиционной калийной селитры для производства особо мощного черного пороха. В 1788 г. на одной из пороховых мельниц собрались: супруги Лавуазье [ ], Бертолле, государственный комиссар Чеврод с дочерью и инженер Лефорт. Смесь хлората калия, серы и древесного угля загрузили в стандартную мельницу для пороха и вся компания отправилась на завтрак. Давно было известно, что совместное растирание в мельницах нитрата калия (калийной селитры), серы и угля вполне безопасно. Дочь Чеврода и молодой инженер Лефорт отделились от остальных и решили вернуться на мельницу. В это время произошел страшный взрыв - и молодые люди, отброшенные далеко от мельницы, погибли. С тех пор было признано недопустимым пытаться заменить селитру в черном порохе хлоратом калия [ 14].
Опубликовано
1. Термин “энтропия” ввел Рудольф Клаузиус в 1865 г. как обозначения превращения энергии в менее ценные формы – “превращение в”
2. Еремин Е.Н. Основы химической термодинамики, 1974, с.72
3. Еремин Е.Н. Основы химической термодинамики, 1974, с.190
4. Современный биограф Людвига Больцмана физик Карло Черчиньяни пишет: “Только хорошо поняв второе начало термодинамики, можно ответить на вопрос, почему вообще возможна жизнь”. В 1906 г. Больцман покончил с собой, поскольку “обманулся в любви; он посвятил свою жизнь атомной теории, но любовь его осталась без взаимности, потому что современники не могли понять масштаб его картины мира” (Шаффер Саймон, Людвиг Больцман и второе начало термодинамики) Интернет: http://if.russ.ru/issue/6/20010816_cha-pr.html
5. Еремин Е.Н. Основы химической термодинамики, 1974, с.193
6. Капра Фритьоф Паутина жизни. Новое научное понимание живых систем. Пер. с англ. – К.: “София”; М.: ИД “Гелиос”, 2002. – 336 c, Глава 5. Модели самоорганизации
7. Энциклопедия для детей: Т.3 (География). – М.: Аванта+, 1994. – 640 с., стр.305
8. Энциклопедия для детей: Т.8. Астрономия. – М.: Аванта+, 1998. – 688 с., стр. 540
9. Гладышев Г.П. Супрамолекулярная термодинамика – ключ к осознанию явления жизни. Москва-Ижевск: Институт компьютерных исследований, 2003, 144 с., с. 27, 125.
10. Впервые введен Массье (1869), затем Дж.Гиббсом в 1875, стал широко известен после статьи Г.Гельмгольца 1882 г.
11. Еремин Е.Н. Основы химической термодинамики, 1974, с.53
12. Краткий справочник физико-химических величин Изд.8-е, Л.: Химия, 1983. – 232 с.
13. Загорский В.В. Огни потешные (Фейерверк: история, теория, практика) М.: "Химия и жизнь - XXI век" 1997 г. - 64 с., илл.; Переиздание: М.: Школа имени А.Н.Колмогорова “Самообразование”, 2000, - 64 с.
14. Marshall Arthur, Explosives. Their manufacture, properties, tests and history, J.& A. Churchill, London, 1915, 624 p., p.25
|
