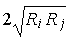 as well), where Ri and Rj are van der Waals radii. These data make it possible to determine the molecular coordination number.
as well), where Ri and Rj are van der Waals radii. These data make it possible to determine the molecular coordination number.
Computer software has been developed for crystal chemistry analysis of arrangement of molecules in organic crystals on the basis of concepts worked out in the studies conducted by P.M.Zorky et al. Initial information is input in the form accepted in Cambridge Structural Database. Partially ordered crystal structures are excluded from consideration.
In calculations and in descriptions of crystal structures rational designations of molecules of the form N–HKL are used, where N is the index of a subsystem of translationally equivalent molecules, H, K, L are crystallographic coordinates of the molecules of the subsystem; it is conventional to use Roman numerals for N. The reference molecule is designated as I–000. Besides, in effect, symbol N–HKL corresponds to a pair of molecules I–000 – N–HKL. Only pairs of adjacent molecules are usually considered; they are called molecule–molecule contacts (or simply contacts, for short) N–HKL. In the case of polysystem crystals, where molecules occupy more than one orbit, symbols N (n)–HKL are used, where n is the index of the orbit.
The standard crystal chemistry analysis includes:1. Revealing the environment of the reference molecule I–000 (in the case of polysystem crystals molecules I(n)–000 are considered to be reference molecules). The list is made of those pairs of molecules for which there is at least one distance rij (atoms i and j belonging to different molecules) that is less than a specified limit R0.
2. Revealing and calculating the shortest atom-atom contacts rij (N–HKL) and their comparison with the values of Ri + Rj (and as well), where Ri and Rj are van der Waals radii. These data make it possible to determine the molecular coordination number.
3. Calculating the energy of contacts N–HKL in atom-atom approximation using 6-exp or 6-12 or some other potentials with or without regard to effective charges on atoms, which are calculated with the help of quantum chemistry methods; the calculation of the total potential energy of the crystal structure (US) and of the energy of molecular agglomerates (Uaggl), i.e. ensembles of the most tightly bonded molecules. Such agglomerates can be islands (dimers, trimers etc), chains, layers, three-dimensional frameworks. The value of Uaggl/US characterizes the efficiency of agglomerates. The determination of the type of the agglomerates that are present in the crystal makes it possible to assign the substance being studied to a particular structural subclass.
4. Constructing various graphic representations of the crystal structure and of its fragments, which clearly demonstrate arrangement of molecules, in particular, constructing representations of the most close molecule-molecule contacts.
 |
 |
|
|
|