Eugene Babaev "Intuitive Chemical Topology Concepts" (c)8. Homeomorphism of Topoids
A topologist, who does not make a difference between the doughnut and the coffee-cup, will never mix them up during a breakfast. Although the objects are homeomorphic, they are functionally not interchangeable, differing in shape and constitution. On the contrary, the homeomorphism of chemical models provides many examples where the homeomorphic objects are, indeed, functionally interchangeable in practice.
Let us look at the homeomorphism of topoids as a continuous mapping of one certain 2D model to another. Topoids are assigned to real molecules with the discrete number of atoms and electron pairs. Therefore, the reverse side of any homeomorphism is discrete (not continuous!) and certain (not arbitrary) leap in the number of atoms and electrons. Equations (11a,b) state that c = 2 N -- Z. Let us fix K = 1 and L = 0. Then, the homeomorphism of molecular topoids (the invariance of c) should follow only from the balance of N and Z. The homeomorphism is expected relating somehow to a chemical similarity. Are there any known types of similarity if only atoms and electrons are counted? An intuitive answer is "Yes": a knowledgeable chemist may easily recall a few famous electron-count concepts, like the Langmuir isosterism, the Hückel rule for aromatic polygons, or the Wade rules for cage boron hydrides.
In the simplest case, the values N and Z may be fixed; then, the homeomorphism (c = const) is expected for isovalent molecules with the same number of atoms. This case corresponds just to the similarity type known as isosterism. Isosteric molecules (like CO and N2, benzene and borazene, POCl3 and SiCl4, etc.) have surprisingly close geometry, spectra, and (quite frequently) physical properties (see reviews [29, 107, 108]).
Moreover, consequent insertion of only certain fragments should preserve the value c. But this is possible only if the inserted fragment has c = 2 N -- Z = 0. Therefore, a topoid of inserted group should be homeomorphic to a torus or cylinder (to which c = 0), and the electron count for this group (2 N = Z) may be written as the ratio Z/N = 2. Consider a set of groups with such a ratio, with small N (from 1 to 4), and with the possibility to insert such a group more than once.
CH2 group. This group (with Z/N = 2 and N = 3) is famous, being responsible for the phenomenon of already mentioned CH2 homology. (The term homology is used here in the chemical sense [116], not in topological [41]; furthermore, chemical homology is an elder concept). Although the principle is very old, it has strong influence on the modern molecular similarity concepts [117], being a sort of reference point to answer the question "What chemical similarity may be at all". Homologues display monotonicity and additivity in physical properties and pronounced similarity in chemical behavior. Sometimes homologues display sort of a periodicity in their properties, that is, superposition of two monotonous functions for the odd and even members. The isosteric combinations -BH2--, -NH2+-, -BH2NH2- may be used to replace -CH2CH2- fragments, but the chains formed are less stable [118].
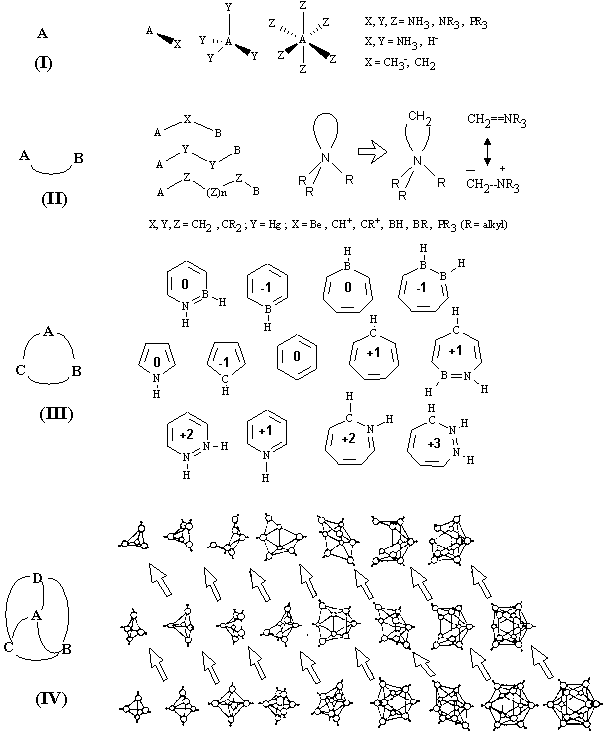
Homeomorphism in CnH2n+x series (see Sections 3.1 and 6.1) resembles the definition of homeomorphic graphs. Expanding the scope on homological relationship, one may insert the CH2 group into any cycle ci, even if a cycle be the lone pair c1. In pseudographs, it looks like the subdivision of a loop. Thus, taking as a precursor a Lewis base (like NR3, PR3, SR2) one may obtain the "homological" structure of a dipolar ylide (like CH2NR3, CH2PR3, CH2SR2) without violation of the genus of a topoid. The ylides are important reagents for organic synthesis [119, 120], used, for instance, in the famous Wittig reaction. The ylides are strong Lewis bases (as are their homeomorphic precursors), although with lone pairs located on the carbon atoms (Figure 23, II). The resonance structures of ylides are frequently drawn as neutral ones with the double bond instead of a lone pair (a sort of intuitive equivalence between the cycles c1 and c2). It is difficult to insert several CH2-groups into the lone pair of NR3 (or into the double bond CH2=NR3), since the pentacoordination is impossible for the nitrogen atom. However, it is do possible for the case of phosphorus, and the -PR3- fragment may belong to a large cycle, resembling CR2 group (R is any alkyl group).
Some cases of CH2 homology, treated as homeomorphism, are featured. The family CnH2n is homeomorphic to the inserted group. Consequently, for the pairs [CH2, CH2], [CH2, CH2=CH2], and [CH2=CH2, CH2=CH2] there is no evidence which group is inserted, and which cycle ci is taken for insertion. The object and the operation with the object become identical. The formation of ylide is also symmetrical: one may treat it as the insertion of -NR3- into methylene’s lone pair, or vice versa; the -PR3- group may be "inserted" into a C-C bond of a large cycle. Furthermore, perhaps in chemistry only, the homeomorphism may be equally "mental and experimental", having parallel to really observed reaction. The abstract insertion of methylene to ethylene corresponds to the high yield reaction, that is, formation of cyclopropanes from carbenes and alkenes. The homology of ammonia and ethylamine (which differ by ethylene) corresponds to Michael addition of amines to alkenes. In any Diels-Alder reaction involving the ethylene and a diene, the resulting cycloadduct is homeomorphic to the initial diene. Even the saturation of ethylene by hydrogen is a specific homological operation (the "insertion" of a CH2CH2 group into an H-H bond). Of course, H2 (a spherical topoid) is the parent for CnH2n+2 series if n = 0. Hence, the chains and cycles may be expanded or shrunk, remaining homeomorphic. Less trivial examples of homological series with delocalized bonds will be discussed in Section 11.
CH+ group. This fragment (with Z/N = 2 and N = 2) is responsible for the similarity in p-isoelectronic chains and cycles. The insertion of CH+ group makes planar delocalized p-systems longer (ethylene to allyl cation or allyl anion to butadiene) without violation of initial planarity and number of p-electrons. This is essential for delocalized cycles. One may easily recall, that 5-, 6-, and 7-membered aromatic rings (cyclopentadienyl anion C5H5-, benzene C6H6, and tropylium cation C7H7+) differ from one another just by a CH+ group [121, 122]. Hence, these "homological" aromatic structures have homeomorphic topoids in quite the same sense as usual CH2 homologs are. Furthermore, the insertion of a CH2 group may also preserve aromaticity, and this phenomenon is known as homoaromaticity [122]. The homological fragment CH+ may be substituted by isosteric groups BH and NH++, the insertion of which into the cycle C5H5- result in the pyridinium cation C5NH6+ and borabenzene anion C5BH6-, both being aromatic and isostructural to benzene. Molecular topoids for aromatic pyrrole and benzene are also homeomorphic (the double bond is substituted by a homeomorphic lone pair), and homeomorphic insertions of BH, CH+, or NH++ groups into the pyrrole ring result in (neutral or charged) heteroaromatic hexagonal structures (see examples in Figure 23, III). Although CH+ cannot form long chains, within the class of fused benzenoid hydrocarbons it may be inserted and removed mutually, resulting in nonbenzenoid arenes. Homeomorphism equally preserves aromatic and antiaromatic types, and the insertion of CH+ (and its isosters) into the antiaromatic rings causes the inheritance of antiaromaticity.
BH group. The fragment BH is essential: it has two vacancies (unlike the methylene group) and the capacity of forming the multicentered bonds with three neighbors (see Section 11). This group, therefore, may serve as a vertex of a polyhedron. Indeed, BH is a well-known homological difference in the families of boron hydrides BnHn+x [123]. Such hydrides form two neutral homological series with x = 4, 6, (nido-, and arachno-boranes, from the Greek for "nest-like", and "web-like", respectively) and one family of dianions [BnHn]2- known as closo-boranes (from the Greek for "closed").
Conventional structures assigned to these three classes with delocalized bonds are remarkable (see Figure 23, IV suggested by Rudolf [124]). The closo-class is represented by "deltahedral" boranes clusters with a skeletal pattern resembling triangulation of the sphere, whereas the nido-, and arachno-classes may be obtained by removal of a cluster vertex in an appropriate closo-polyhedron (deltahedron) [124 -- 127]. An attempt to calculate their Euler characteristic using formula (11) results in x = c (just as it was for the CnH2n+x family). The values for BnHn+4 (c = 4) and BnHn+4 (c = 6) clearly indicate that the genus exceeds the value allowed to the closed orientable 2D manifolds (maximum c = 2 for sphere), and the nature of their topoids will be discussed in Section 12.
Already mentioned CH+ group may be also inserted into the closo-polyhedron as another homological fragment, resulting in the stable carborane families [CBnH2n]- and C2BnH2n [111] with preservation of the closo-structure. Of course, the Euler characteristic c = --2 is the same for every class. Hence, the topoid should be a pretzel, and the graph should be bicyclic. However, only the first member of the neutral carboranes matches such a structure (three BH fragments inserted into the bonds of acetylene pretzel result just in C2B3H6), whereas higher homologs are delocalized. Other examples of CH+-homology in delocalized clusters are represented by the polyhedrons C5H5+ and C6H62+. Hence, we may conclude that insertion and removal of the homological BH and CH+ groups in such clusters is equivalent to an inessential perturbation of a structural pattern around the cage.
NH3 group. Unlike other above-mentioned homological groups, the group NH3 (with Z/N = 2 and N = 4) cannot subdivide chains, cycles, or clusters. This group, however, is able to make a shell around a point-like cation. Indeed, ammonia is a common ligand in coordination chemistry, capable of multiple coordinating to either a transition metal cation or even to a main-group cation (like in complexes M(NH3)nm+ in liquid ammonia [128]). One would say that such a coordination of several ligands to a Lewis acid makes no essential change to the initial cation (Figure 23, I). Perhaps, only the total size and shape are increased upon the coordination. The size and shape, however, are geometrical properties, not topological, and the coordination resembles a remarkable homeomorphism from a sphere to the 2D surface of a hedgehog.
The same homeomorphism (with more pronounced enlarging of the spherical shape) is typical for ligands with longer chains, the homologs of ammonia (like mono-, di-, and trialkylamines). The formed external shell becomes hydrophobic. The higher hydrophobicity (and higher stability) of such complexes may be achieved by the addition of novel handles to the spherical surface. Thus, the use of cyclic polydentate ligands (like kryptands and aza-analogs of crown ethers) results in so stable hydrophobic cations, that the initial mineral salt may be easily dissolved even in a nonpolar solvent. The topology of such complexes, described in terms of graphs (trees and polycycles) or surfaces (sphere with pasted handles) is completely parallel to the picture of saturated hydrocarbons CnH2n+x. The molecules PR3 (isovalent and homeomorphic to NH3) represent another class of suitable ligands, as well as the homeomorphic carbenes CR2, which may form complexes with transition metals. Topological shrinking of ligands (removal of NH2+ group from NH3 or CH+ group from CH2) results in the hydride anion H-, which is another common ligand capable for coordination.
Figure 23. Design of molecular structures by the insertion of certain groups with c = 0 (see the text). (I) The use of ligands in coordination chemistry (A is a center with vacancies). (II) Examples of groups that may be inserted into a bond A–B without changes of the genus. (III) Insertion of groups BH, CH+, NH2+ into the planar aromatic rings with preservation of aromaticity (the number within the ring is the charge of molecule). (IV) Insertion of groups BH and CH+ within the series of boron hydrides and carboranes with preservation of structural pattern (isostructural families are in horizontal rows). Arrows indicate the relationship between arachno- (top row), nido- (middle row) and closo- (bottom row) structures.
A special case, that (with some care) may be treated as a sort of homeomorphism, is the insertion of metals from the second column of the Periodic Table with Z/N = 2 (Mg, Zn, Hg, and Cd) into some bonds (e.g., C–Hal or Hal–Hal), although only a Hg atom may form short chains. These reactions are extremely useful in organometallic chemistry (e.g., the Grignard reaction), and homeomorphic structures are formally enlarged without change in their topology. Here the simplest "homologs" (with pronounced ionicity of bonds) dramatically differ from the parent members (with covalent bonds), however, there are still not enough data to conclude about the similarity types within longer homological series (with several metal-metal bonds).
Let us emphasize that the concept of homeomorphism of topoids, being applied to the inserted fragments, brings together many previously disjoint chemical similarity types in a unique manner. Although the enlarging or diminishing of an object (a geometrical change) appears in homeomorphic series, the initial topological pattern is conserved. Furthermore, we may even classify the initial patterns (and their homeomorphisms) in terms of usual topological cells:
e0 cell (a point-like cation) homeomorphism around the point (Figure 23, I);e1 cell: (a linear bond) homeomorphism along the line (Figure 23, II);
e2 cell: (a fragment of planar cycle) homeomorphism around the plane (Figure 23, III);
e3 cell: (a spatial cavity in deltahedrons) homeomorphism around the space (Figure 23, IV).
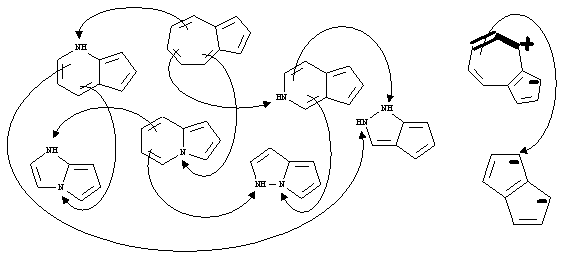
The inserted groups only slightly overlap, each being responsible for its own cell and dimension. The evident homeomorphism between groups indicates that homeomorphism in chemistry is just a concept independent of dimensions.Aromaticity and homeomorphism. Note that the total number of handles is not responsible for aromaticity or antiaromaticity, expressed by the Hückel rule (4n and 4n+2 p-electrons). The presence of cycles ci (i>2) and their planarity is essential, because isolated handle (a multiple bond or a lone pair) outside the delocalized perimeter brings nothing to the p -electron count. However, the possibility to count p electrons appears only because there are small cycles c1 and c2 within the perimeter of a cycle ci. Therefore, for the case of usual polycyclic conjugated hydrocarbons with total C’ large cycles ci (i>2) and total C handles in topoids (C’ plus double bonds) the difference between "large and small" handles C-C’ corresponds just to the Hückel rule: it is odd for the aromatic case and even for antiaromatic. Of course, the groups BH, CH+, and NH+ may be freely inserted or removed without changing the C-C’ value. This rule is also not violated if the cycles c1 of NH groups are counted as handles. Moreover, since the fragments NH, CH-, BH2-, CH=NH+, and CH=CHCH+ have the same topology of a pretzel (intact or bitten) and bring two p-electrons, they may be freely interchanged without loss of aromaticity (or antiaromaticity). This adds real "flexibility" to the common (hetero)aromatic structures, because one may topologically shrink larger fragments to smaller ones (see examples in Figure 24).
Figure 24. Topological shrinking and preservation of aromaticity. On the left: Design of heteroaromatic pseudoazulenes from azulene structure as a shrinking of double bond(s) to lone pair(s). On the right: Shrinking of azulene (shown as resonance form) to dianion of pentalene as homeomorphism of allyl cation (bold fragment) to p-isoelectronic CH3-anion.