Введение
По отношению к естественным наукам математика была и остается недостижимым образцом точности, к которому стремится любая наука, изучающая природные явления. Одна из ключевых особенностей математического мышления заключается в выделении идеальных множеств, замкнутых относительно какой-либо операции изменения самих объектов этого множества. Так, можно рассматривать множества чисел, матриц или многочленов как некоторые множества объектов, на которых определяются операции (различные типы сложения, умножения и т. д.), не выводящие за пределы исходных множеств. Примерами математических структур, определяемых именно таким образом, могут служить алгебры, группы, кольца, поля и т.д., порождающие богатый арсенал математических объектов.
Замкнутые математические структуры играют огромную роль в естествознании. Частный случай таких структур — группы — формализуют качественное представление о симметрии объектов, выражая в символьной форме интуитивные представления о соразмерности, гармонии и порядке частей целого. Хорошо известные приложения теории групп в химии [1,2] ясно показывают, какую именно пользу может дать анализ симметрии волновых функций или симметрии размещения атомов в молекуле как точек пространства. Между тем, применение идей теории групп в физике дает пример принципиально иной возможности использования аппарата замкнутых множеств в естествознании. Физики давно заметили, что наличие у объекта некоторой симметрии, описываемой замкнутой групповой структурой, равносильно наличию некоторого (явного или скрытого) свойства инвариантности, т. е. закона сохранения.
За последнее столетие химия накопила богатый арсенал алгебраических моделей, успешно используемых для решения задач типа "структура—свойство"применяемых в компьютерном синтезе или нацеленных на выяснение степени молекулярного подобия (см., например сборники [2—4] и библиографический обзор [5]). Заметим, однако, что среди известных алгебраических моделей нам неизвестно ни одного примера, когда такая модель привела бы к формулировке какого-либо нового принципа сохранения. Можно сказать, что химия вообще чрезвычайно бедна на инварианты и законы сохранения. Так, если отбросить инвариантность массы, заряда и энергии (физические по своей сути законы сохранения), то останутся, разве что, правило сохранения орбитальной симметрии [6] и принцип инвариантности Эйлеровой характеристики [7].
В настоящей работе мы попытаемся найти пример замкнутого множества там, где его ранее не искали или не замечали. Мы покажем, что среди привычных взгляду химика-органика структур можно математически строго выделить класс молекул, обладающих определенным “ритмическим” подобием электронного строения, связанным с альтернированием полярных (донорных и акцепторных) центров. В качестве операции изменения таких объектов (называемых ниже суперконсонантными) мы будем рассматривать обычные полярные реакции, включающие стадии гетеролитического образования и/или разрыва связей. Основная идея настоящего подхода заключается в том, что указанное множество, при определенных допущениях, действительно можно считать “почти замкнутым” относительно указанной операции изменения. В свою очередь, такое утверждение равносильно новому и нетривиальному для органической химии принципу сохранения топологического свойства полярной двудольности (зарядового альтернирования) в полярных процессах. Формулировке модели предшествует обзор ранних работ, посвященных принципу альтернирования и проблеме консонантности, до сих пор никак не нашедший отражения в отечественной литературе.1. Принцип альтернирования: история и современное состояние проблемы
Кажущаяся универсальность в сочетании с расплывчатостью формулировок делали модель Лэпворта весьма уязвимой, что породило продолжительную полемику между Лэпвортом и Робинсоном, с одной стороны, и Ингольдом, с другой. Ряд факторов казался противоречащим правилу, что пытался продемонстрировать Ингольд в серии работ под названием “Природа эффекта альтернирования в углеродной цепи” [10], направленных на опровержение этой модели. В связи с тем, что Ингольд неправильно интерпретировал исходные принципы модели, а к тому же сам допустил ряд ошибок (в том числе экспериментальных), дискуссия приняла весьма острый и эмоциональный характер [11]. В 1926 г. Английское Королевское Общество нашло весьма оригинальный выход из ситуации, наложив своеобразное вето на продолжение полемики, назвав ее “играми в химические крестики и нолики”, и отказываясь “впредь рассматривать и публиковать статьи, посвященные мистике полярностей” [12]. Прекратив дискуссию, Лэпворт и Робинсон фактически приостановили и дальнейшую разработку модели, тогда как язык электронных смещений, введенный Ингольдом, целиком вытеснил терминологию альтернирования. Материалы историков науки по этому вопросу опубликованы сравнительно недавно [13, 14], а впервые внимание химиков к имени Лэпворта было привлечено [15] лишь в 1972 г. (через год после смерти Ингольда).
Заметим, что фактический запрет на дальнейшее развитие идеи на Британских островах не ослабил интереса к ней мирового сообщества. Наряду с Лэпвортом, например, сходные идеи высказывали другие европейские [16, 17] и американские химики [18, 19]. Ряд учебников вплоть до середины века упоминал принцип альтернирования не только с критической точки зрения, но и как полезную теоретическую модель [20—22]. Ясно, что понятия мезомерии и альтернирования глубоко синонимичны и в равной мере приемлемы для описания перераспределения электронной плотности в пи-системах с гетероатомами. Тем не менее, большинство данных подтверждало введенную Ингольдом модель затухания эффекта полярной группы вдоль насыщенной цепи.
Между тем, притягательная парадигма альтернирования, как своеобразная “идея-призрак”, продолжала переоткрываться заново экспериментаторами, эпизодически возникая и исчезая из поля зрения теоретиков. Особое внимание привлекал [19, 23—26] и продолжает привлекать [27—32] экспериментально наблюдаемый для отдельных классов эффект альтернирования в насыщенной цепи, подтверждаемый как спектральными, так и кинетическими исследованиями. В ряде случаев, однако, ожидаемый эффект обнаружен не был [33, 34]. Мы не будем останавливаться на известных примерах альтернирования физических свойств [35], типичных, например, в ряду алканов [36].
Разумеется, идея альтернирования полярностей не осталась без внимания со стороны квантовой химии. Чередование зарядов в альтернантных пи-системах с гетероатомами проявляется даже в простейшем методе МОХ. Менее тривиально появление заряда противоположного знака на центральном атоме в аллильном катионе и анионе, проявляющееся при использовании теории ВМО [37]. Своеобразный ренессанс принципа альтернирования в 70-х гг. можно связать с появлением работы Попла и Гордона [38], когда первое тестирование полуэмпирического метода CNDO/2 на широком классе структур показало явное альтернирование заряда, в том числе в насыщенных цепях. Позднее этот же результат был подтвержден расчетами по методу ab initio [39], см. также критический анализ и библиографию в работе [40]. Возможно, с физической точки зрения, причины феномена более глубоки [41].
Интересные результаты получены при анализе принципа альтернирования с термодинамической точки зрения. Термохимические данные свидетельствуют [42], что для малых молекул соседство однотипных по полярности функций возле одного атома (т. е. пары доноров или пары акцепторов, приводящее к альтернирующей последовательности) энергетически более предпочтительно, чем соседство функций противоположной природы (т. е. с нарушением альтернирования), что отражается в направленности реакций диспропорционирования. Аналогичным образом, стабильность замещенных олефинов или бензолов определяется, в первую очередь, альтернирующим или неальтернирующим окружением двойной связи (бензольного ядра) полярными группами. Следствием этого правила, в частности, оказывается [43] неожиданно более высокая стабильность кросс-сопряженных фрагментов над системами с обычным сопряжением, особенно ярко проявляющаяся в случае карбанионов.
Заслуживают внимания опубликованные недавно обзор и монография Хо — известного американского химика-синтетика [44, 45]. В развиваемом им подходе делается попытка использовать принцип альтернирования в качестве инструмента для объяснения самых разнообразных фактов реакционной способности и региоселективности в органической химии на основе допущения, что альтернирующее расположение функций вокруг атома всегда более энергетически предпочтительнее, чем при отсутствии такого альтернирования. Обсуждаемый круг фактов включает, например, проблему активации субстрата (путем как усиления, так и нарушения альтернирования), объяснение наблюдаемой региоселективности в процессах замещения, окисления, присоединения и циклоприсоединения, движущую силу перегруппировок. Сам автор предлагает рассматривать принцип альтернирования как удобное мнемоническое правило, позволяющее решать конкретные задачи.
1.2. Принцип альтернирования и планирование синтеза. По-видимому, наибольшее число приложений принцип альтернирования нашел в области планирования органического синтеза. Начало было положено работой Кори [46], в которой вводилась операция ретросинтеза, т. е. мысленного гетеролитического расчленения целевой структуры на условные субъединицы синтонов. Такое расчленение определялось как “логичное” или “нелогичное” (logical and illogical disconnection) в зависимости от того, насколько полярная структура образующегося синтона соответствует привычному распределению полярностей в структуре реального реагента (ср. “логичный” ацилий-катион и “нелогичный” ацильный анион). От этого определения оставался всего шаг до анализа взаимосвязей между альтернированием полярностей в целевой структуре с таковым альтернированием в ее предшественниках.
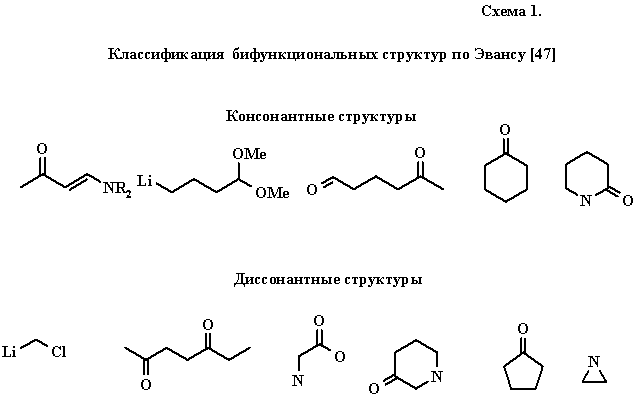
В мае 1971 г. на университетском семинаре Д. А. Эванс сделал доклад [47], оказавший решающее влияние на формирование целого ряда математических моделей органического синтеза. Рукопись Эванса, разосланная многим химикам, так никогда и не была опубликована, тем не менее, на этот доклад неоднократно ссылались (см. например, [48, 49], а также подробный комментарий на каталонском языке [50]). В этой работе Эванс предложил простую и элегантную классификацию бифункциональных (т. е. содержащих две полярных функции) молекул на консонантные и диссонантные. Консонантные структуры допускают разметку углеродных атомов таким образом, что они напоминают заранее заданный шаблон (charge affinity pattern) с альтернирующей последовательностью электро- и нуклеофильных центров. В диссонантных молекулах такое представление невозможно (схема 1).
Заметим, что принципиальных ограничений на насыщенность цепи (как и в модели Лэпворта) не налагалось. По-видимому, Эванс одним из первых сформулировал математически строго, как именно можно добиться максимального использования уже имеющихся в молекуле функциональностей при планировании ее синтеза, заметив, что синтез консонантной структуры (или консонантной цепи сложной молекулы) удобно осуществлять из консонантных же предшественников.
Существенно, что у Эванса в качестве концевых групп бифункциональной молекулы или шаблона может выступать не только электроотрицательный гетероатом (N, О, F как у Лэпворта), но и элементоорганический фрагмент (с металлом 1 или 2 группы, алюминием или кремнием), имеющий противоположную поляризацию центров в цепи. В специальный класс выделялись нитрогруппа и функции, содержащие серу, фосфор, бор и другие элементы, которые приводили к неоднозначному распределению полярностей в цепи, делая один и тот же атом одновременно электро- и нуклеофильным. В экспериментальных работах Эванса [51] обсуждались другие способы, позволяющие инвертировать исходную, консонантность альтернирующей цепи.
Несколько позднее появился известный обзор Зеебаха [52], в котором (со ссылкой на частное сообщение Эванса) вводилась более упрощенная и альтернативная терминология разделения полярных структур на “нормальные” (т. е. с альтернирующей последовательностью донорно-акцепторных центров в цепи) и структуры, содержащие “умполунг” (инверсию полярности какого-либо донорного или акцепторного углеродного центра относительно гетероатома на конце цепи). Ограничиваясь рассмотрением в качестве гетероатомов лишь азота и кислорода, Зеебах вскользь упоминал о синтезе структур нормального строения из других нормальных,предшественников (реакциями типа альдольной или кляйзеновской конденсаций, реакций Принса, Манниха и Михаэля), считая эту закономерность своеобразным синтетическим ограничением.
Главной заслугой Зеебаха является детальный анализ методов (ряд которых был впервые предложен его исследовательской группой), с помощью которых можно создать умполунг. Заметим, что в этот термин с самого начала закладывается двойной смысл: умполунг как структурная особенность (например, четность цепи между парой гетероатомов) и умполунг как процесс, при котором донорная или акцепторная природа атома инвертируется. Для нашего дальнейшего анализа принципиально, какие именно методы позволяют создать умполунг. По мнению Зеебаха следует различать 6 таких способов: (1) 1,2N-окисление, (2) обмен и модификация гетероатома, (3) гомологизация и ее обращение, (4) использование циклопропанов, (5) использование ацетиленов и (6) редокс-реакции. Разумеется, исходными реагентами по отношению ко всем этим методам выступают структуры нормального строения. В качестве предельного случая умполунга как структурной особенности (ни один из 6 методов не нужен) формулируется понятие прямого умполунга (например, в оксиде углерода, изонитрилах и т. д.). Наконец, рассматривается обращение умполунга как процесс, приводящий к структурам нормального строения.
Не случайно, что дихотомическая классификация структур и реагентов на консонантные и диссонантные (или нормальные и с умполунгом), построенная на чисто математическом определении, привлекла внимание специалистов в области компьютерного синтеза. Первая из программ TOSCA разрабатывалась специалистами немецкого концерна Hoechst AG в течение 5 лет [49] и привела к предсказанию новых путей синтеза искусственных подсластителей с консонантной структурой. Подробная формализация перехода от консонантных синтонов к реальным структурам реагентов используется в программе STRATOS [53]. Наконец, третья компьютерная программа CHAOS [54], основанная на анализе путей синтеза бифункциональных молекул, зарекомендовала себя как удобное средство для обучения принципам компьютерного синтеза.
Используя модель альтернирования для создания компьютерных программ, авторы вносили уточнения и более четко очерчивали область применимости исходной модели. В особенности это относится к работе [49], где подчеркивалось, что наибольшие перспективы модель консонантных и диссонантных реагентов могла бы найти в области гетероциклического синтеза, где полярные процессы (например, циклоконденсации) играют ключевую роль. Заметим, что внутри самой химии гетероциклов важность принципа альтернирования для планирования гетероциклического синтеза осознана явно недостаточно. Единственный раз правило альтернирования было вскользь упомянуто недавно авторами обзора [55] при попытке обобщить данные по методам конструирования шестичленных гетаренов.
Такая ситуация побудила автора настоящей работы провести детальный анализ типов полярной структуры реагентов, когда-либо использовавшихся для конструирования пиридинового ядра. Результаты анализа литературы позволили создать компьютерную базу данных [56, 57], обработка которой привела к заключению, что в 95% случаев соблюдается магическое правило "структура— синтез" [58], а именно, альтернирующая полярность центров пиридинового ядра жестко детерминирует полярность ациклических реагентов. Лишь в единичных случаях для синтеза цикла использовались реагенты с умполунгом (часто с побочным образованием пирролов) или протекал процесс обращения умполунга. Это правило (строго соблюдавшееся также для хинолинов) было положено в основу компьютерной программы Heterocycland [59], позволявшей перечислять полярные типы реагентов для синтеза гетаренов и прогнозировать неизвестные ранее пути синтеза. Экспериментальное подтверждение модели было осуществлено в новом, ранее неизвестном синтезе хинолинов [60].
2. Критические замечания и необходимость дальнейшего развития парадигмы альтернирования
Проведенный анализ литературы по проблеме альтернирования ясно показывает существование значимого феномена, обнаруживаемого в весьма далеких областях как теоретической, так и экспериментальной химии. К сожалению, нередкое отсутствие взаимных ссылок у авторов подходов затрудняет разработку единой терминологии. Авторы программ, например, предпочитали описывать структуру молекул в терминах консонантности и диссонантности, используя термин умполунг именно для описания реакций. (Дополнительно усложняет языковую проблему терминология, введенная Хо [45], который предложил для консонантных и диссонантных структур труднопереводимые термины “conjoint” и "disjoint".) Поскольку “музыкально-лингвистические” термины консонантность и диссонантность, на наш взгляд, наиболее легко формализуемы, мы также будем следовать терминологии Эванса.
Рассмотрим теперь, какие именно аспекты проблемы альтернирования представляются не до конца прояснёнными и заслуживают дальнейшей разработки. Во-первых, не вполне ясно, следует ли считать консонантность локальным свойством атома (например, обусловленным его окружением), локальным свойством цепи или фрагмента (бифункциональное отношение между парой полярных функций) или глобальным свойством полифункциональной молекулы как целого.Второй, весьма серьезный аспект проблемы — игнорирование или пренебрежение степенью ненасыщенности цепи. Начиная с Лэпворта утверждалось, что эффект альтернирования наиболее сильно проявляется в сопряженной цепи; тем не менее, попытки обобщить приложимость эффекта для цепей частично ненасыщенных или полностью насыщенных не прекращаются до сих пор. Известно, однако, что в насыщенных цепях с полярной группой на конце конкуренция между эффектом альтернирования и эффектом затухания разрешается в пользу последнего. Максимум, что удается зарегистрировать экспериментально — пилообразную зависимость эффекта альтернирования, затухающую примерно у пятого звена насыщенной цепи [23—32]. Аналогичным образом, при планировании синтеза достаточно длинной (пусть консонантной, но насыщенной) цепи выбор между консонантными или диссонантными предшественниками может не являться принципиальным.
Третий вопрос, практически не затрагиваемый в цитируемых подходах, можно сформулировать так: действительно ли для проявления альтернирования необходим мощный (электроположительный или электроотрицательный) неуглеродный заместитель. Химия полна примеров, когда выраженный полярный эффект проявляет, например, алкильная, алкенильная или алкинильная группа. Вместе с тем, заряженные углеродные фрагменты (карбокатионы и карбанионы), т. е. синтоны без гетероатомов — реальность современного органического синтеза. Цитировавшийся выше обзор Клейна [43] свидетельствует о том, что правило альтернирования имеет перспективы приложения в области малополярных пи-систем.
Возможный подход, который позволил бы ответить на поставленные вопросы, должен каким-либо образом учитывать полярность связей С—Н. Заметим, что этот вопрос поставил еще Лэпворт, считавший, что водород можно условно считать электроположительным атомом, равноправным с другими “ключевыми” гетероатомами. Предвосхищая модель консонантных и диссонантных структур, Лэпворт предлагал рассматривать “гомогенные” и “гетерогенные” [8] способы взаимного расположения водорода и гетероатомов в цепи (см. также комментарий Робинсона [9] по этому поводу). Так, уксусная кислота — гомогенная структура, так как допускает однозначную разметку атомов знаками плюс и минус, тогда как муравьиная кислота — гетерогенна и может быть размечена двумя способами в зависимости от того, какой атом — водород или кислород — принять ключевым(схема 2). Лэпворт ограничился самой возможностью введения такой классификации и более не возвращался к ее развитию.
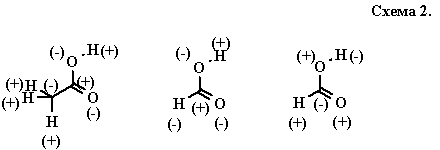
3. Принцип суперконсонантности.
Назовем некоторую цепь между произвольной парой ключевых атомов в структуре гомогенной, если:Будем считать частным случаем нечетной цепи одноуглеродный фрагмент и включим в определение четной цепи ее отсутствие (т. е. прямое соседство функций). Разумеется, все атомы углерода гомогенной цепи допускают строго однозначную разметку чередующимися знаками (скажем, теми же плюсами и минусами).
- между любой парой гетероатомов цепь нечетна;
- между любым из гетероатомов (N, О, F) и атомом водорода цепь четна;
- между любой парой водородных атомов цепь нечетна.
Определение 1. Молекулы, построенные только из гомогенных цепей, будем называть гомогенными.Очевидно, что большую цепь можно составить из меньших, а также представить цикл как замкнутую цепь. Следовательно, можно включить в рассмотрение циклы (в том числе сочлененные) или структуры, содержащие гетероатом внутри цепи (в частности, гетероциклы). Существуют два пути конструирования множества таких гомогенных молекул. Первый путь — аналитический: имея бесчисленное множество структур, мы устанавливаем фильтр на основе определения 1, отделяя гомогенные структуры от тех, которые таковыми не являются. Второй путь — конструктивная генерация гомогенных структур на основе наращивания на произвольную (насыщенную или ненасыщенную) цепь новых атомов, так чтобы возникали исключительно гомогенные цепи (циклы).
Из сопоставления гомогенных структур с консонантными видно, что эти множества пересекаются лишь частично. Так, трет-бутанол, ацетон, уксусная и угольная кислоты гомогенны и консонантны, тогда как метанол, формальдегид, ацетальдегид или муравьиная кислота консонантны, но не гомогенны. Вместе с тем, к числу гомогенных структур придется в силу определения отнести малополярные или неполярные структуры типа пропина, изобутилена или неопентана, вообще не описываемые в терминах консонантности.
Полученная путем доведения до логичного конца модель Лэпворта оставляет, однако, некоторое чувство неудовлетворения, не отвечая на все вопросы, поставленные выше в разделе 2. Во-первых, в определение не было введено представление о заряженных частицах, в результате чего приходится формально отождествлять изоструктурные катионы и анионы, принципиально различающиеся своей полярностью и реакционной способностью (ср. “нелогичный” ацильный анион и “логичный" ацилий катион, электрофильные ОН или NН2-катионы и нуклеофильные гидроксил- и амид-анионы). Наконец, в модели никак не различаются карбеноидные (метилен, дифторметилен, нитрен) и некарбеновые частицы. Итак, необходимо, чтобы в классификацию в явном виде включалось представление о взаимном расположении центров Льюисовской кислотности и основности, т.е. о вакансиях и неподеленных парах. Рассмотрим, каким путем можно достигнуть требуемого результата.
3.2. Обобщение модели полярной гомогенности для ионов. Для описания передачи некоторых полярных эффектов водорода или гетероатома химики давно придумали удобный методический прием: мысленная гетеролитическая диссоциация связи с образованием предельной (несвязной) резонансной структуры из двух ионов. Таким путем, например, можно расщепить связь С—Н в метильной группе изобутилена (на протон и карбанионную часть) или связь С—F в перфторизобутилене (на фторид-ион и карбокатионную часть) для наглядного объяснения эффекта метильной или трифторметильной группы. Формально, ничто не мешает повторять эту процедуру многократно, до расчленения ковалентной структуры на многократно заряженный углеродный остов, окруженный ансамблем внешних ионов (в нашем примере — протонов илифторид-ионов), см. схему 3. В общем случае можно применить эту операцию и для гетероатомов типа кислорода и азота (порождая предельные структуры оксид- и нитрид-иона).
В случае подобного расчленения гомогенных структур образующийся углеродный остов обладает замечательной особенностью. По определению, атомы углерода в таких структурах уже имеют метки плюс и минус, а при отщеплении ионов локализуемый заряд имеет тот же знак, что и исходная метка. Поскольку вновь порождается альтернирующая структура, ничто не мешает довести процедуру ионного расчленения до логического конца, гетеролитически разрывая связи С—С с образованием предельных ионов С4+ и С4- (см. схему 3).
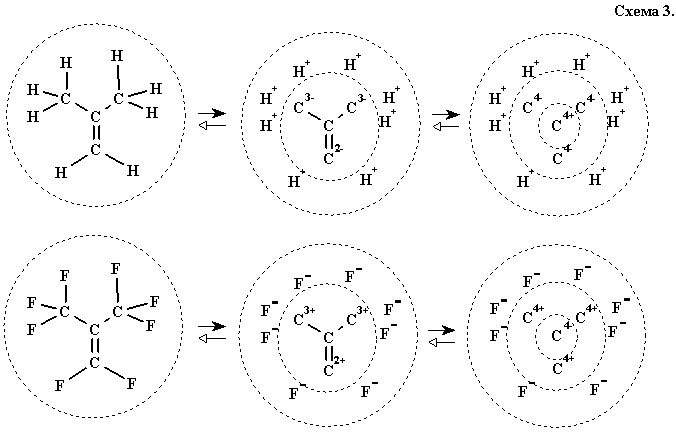
Иными словами, справедлива следующая замечательная теорема 1:
любую гомогенную молекулу можно мысленно разобрать на альтернирующую совокупность ионов (С4+ Н+) и (С4-, N3-, О2-, F-).
Очевидно, что негомогенную молекулу представить таким образом нельзя — обязательно появится "лишний" ион кроме 6 перечисленных (ср. структуры гидразина, этана или метиламина). Вместе с тем, процедуру расчленения на ионы можно инвертировать и рассмотреть последовательную сборку некоторой молекулы из указанной совокупности ионов, причем так, чтобы соседствовали всегда катион и анион. Можно полагать, что после подобной сборки произойдет реорганизация гипотетических ионных связей в обычные ковалентные (полярные или неполярные) связи необходимой кратности в соответствии с обычными правилами валентности. Нетрудно доказать, что при этом будет справедлива следующая (обратная) теорема 2:
любую гомогенную молекулу можно мысленно собрать из запаса ионов (С4+, Н+) и (С4-, N3-, О2-, F-), причем так, что каждый катион соединяется с анионом.Таким образом, можно конструктивно генерировать полярно гомогенные структуры не только исходя из четности цепей между “ключевыми” атомами, отводя углеродным атомам пассивную роль, но и, чисто формально, исходить из представления о двух — катионоидном и анионоидном — атомах углерода как равноправных субъединицах гомогенных структур. Между тем, совершенно необязательным становится неявное требование электронейтральности результирующей структуры: можно остановиться на произвольном моно- или полизаряженном ионе или цвиттер-ионе. Полученное множество является уже неким качественно новым объектом, для которого можно предложить новое название.
Определение 2. Молекулы и ионы, которые можно мысленно разобрать исключительно на ионы (С4+, Н+) и (С4-, N3-, О2-, F-) или собрать из этих ионов так, чтобы каждый катион соединялся с анионом будем называть сильно консонантными или суперконсонантными. (Эти же термины будем использовать и для исходного запаса ионов.)
Математик мог бы сказать, что этим определением вводится некоторое отношение эквивалентности на множестве молекулярных структур. Действительно, любая молекула строго является либо суперконсонантной (и тогда эквивалентна другим таким же структурам), либо нет.
Представление о ковалентных молекулах как ансамблях ионов имеет давнюю
традицию: именно так в прошлом пытался представить себе картину передачи
влияния заместителей Робинсон [9] и именно так, включая два типа углеродных
ионов, рисовал себе картину альтернирования Фаянс [19]. Проблема состояла
лишь в том, что этому принципу придавали слишком широкое толкование и использовали
в тех случаях, где он, возможно,
малоприменим. (По поводу критики представлений Робинсона об “усилении и
дезинтеграции октетов” вдоль альтернирующей цепи см. [20]). Наша задача
в другом: дав строгое определение некоторого класса, выяснить качественные
особенности электронного строения, реакционной способности и методов синтеза
структур этого класса.
4.1. Главные особенности. Первая чисто топологическая особенность суперконсонантных структур состоит в том, что любая такая структура описывается двудольным графом. Как известно из теории графов [61], граф называется двудольным, если множество его вершин можно разбить на два непересекающихся подмножества (скажем, разного цвета) так, что ребро в графе соединяет обязательно вершины разного цвета. Двудольность графа автоматически исключает нечетные циклы. Пользуясь ранее введенной нами символикой [58, 62] соотносить цвет с зарядом (“фильностью”), условимся графически представлять белой вершиной в графе катионоидный центр, а черной — анионоидный. Ниже мы покажем, что двудольность порождает весьма элегантную генетическую взаимосвязь внутри класса суперконсонантных структур.
Вторая особенность относится к взаимному расположению реально заряженных центров, неподеленных пар и гетероатомов. В простейших случаях карбокатионный центр таких структур, по определению, окружен атомами с неподеленными парами, а карбанионный центр — электроположительными углеродсодержащими фрагментами типа ацетил-, карбокси- или циано-групп, что охватывает случаи мезомерной стабилизации заряда. С другой стороны, пример индуктивной стабилизации иона (карбокатиона — метильной, а карбаниона — трифторметильной) также можно считать характерной особенностью суперконсонантных структур. Типичные стабилизированные ионы — знакомые трет-бутильный катион или гуанидиний, анионы типичных СН-кислот, перфтор-трет-бутильный и карбонат-анионы.
Цианид или ацетиленид анионы, как и простейший метильный катион, очевидно, противоречат определению суперконсонантности. Также понятно, что с этим определением несовместим карбеновый или нитреновый тип структуры. Наконец, ион аммония или гидроксония подходит под данное определение, тогда как тетраметиламмоний или триметилоксоний — нет.Последнее, почти очевидное свойство, состоит в том, что если структура суперконсонантна, то сумма ее валентных электронов всегда кратна восьми (обратное утверждение неверно). Это автоматически следует из представимости таких структур октетными анионами (вносят по 8 электронов) и катионами С4+ и Н+ без валентных электронов.
4.2. “Суперконсонантный мир”. Рассмотрим подробнее структурные фрагменты, которые можно встретить среди суперконсонантных молекул. Подчеркнем, что в силу формальной ионной аналогии, вытекающей из определения 2, любую такую молекулу можно рассматривать как своеобразную “неудавшуюся соль”. Знакомство с такими структурами можно скорее назвать припоминанием: слишком много тривиальных (или имеющих тривиальные названия) соединений подпадают под определение 2. Легко видеть, что это множество включает, например, ацетон и продукты его олигоконденсации (оксид мезитила, форон, изофорон, мезитилен), классические СН-кислоты (малоновая, ацетоуксусная, ацетондикарбоновая кислоты и их нитрилы, ацетилацетон, димедон, кислота Мельдрума) и их енольные формы, или енамины, кетен и его димер, угольную кислоту и недокись углерода (С3О2).
Среди ароматических структур сильно консонантными являются, например, орсин, флороглюцин, симм-ксилидин, кислоты типа орселиновой, гирофоровой или леканоровой. Многообразны азотсодержащие структуры с фрагментами цианамида, мочевины, гуанидина. Среди гетероциклов можно упомянуть имид Гуареши, 2,4,6-триметилпириллий, симм-коллидин, дегидрацетовую, барбитуровую и циануровую кислоты, триацетонамин.
Ясно, что двудольность (запрет нечетности цикла) ограничивает суперконсонантные структуры четно-циклическими классами, например, замещенными бета- или дельта-лактонами, циклобутанами и циклогексанами, адамантанами или кубанами. Что касается типов цепей незамкнутых структур, то характерными примерами будут являться насыщенная цепь полиизобутилена, сопряженная полиеновая цепь с альтернирующим расположением донора и протона (или донора и акцептора), а также полииновая или (нечетная) поликумуленовая цепи.
Заметим, что три существенных ограничения препятствуют полноценной реализации суперконсонантного мотива. Во-первых, это понимаемая в глобальном смысле таутомерия, препятствующая геминальному взаиморасположению кислых функций (ср. гипотетическую ортоугольную кислоту, гидрат ацетона и аналогичные гем-производные), а также соседству протонодонорной функции с кратной связью (гидрокси- или аминоацетилен, простейшие енолы). Во-вторых, это стерический эффект сильно разветвленных суперконсонантных фрагментов типа неопентильной или трет-бутильной групп (ср. три(трет-бутил)амин, тетра(трет-бутил)метан и т. д.). В третьих, это факторы, влияющие на стабилизацию пи-систем (так, суперконсонантный 1,3-диметилциклобутадиен антиароматичен). Отметим, что стабилизирующее ароматическое воздействие может согласовываться с суперконсонантным мотивом (в случае подходящим образом замещенных бензолов, нафталинов и т. д.) или противоречить ему. Так, известные дианионы 1,3-диметилциклобутадиена или 1,3,5,7-тетраметилциклооктатетраена альтернантны и ароматичны, но тем не менее, неразбиваемы на суперконсонантные ионы.
4.3. Генетические отношения в ряду суперконсонантных структур. Отмеченное структурное многообразие сильно консонантных молекул не противоречит, однако, сформулированному выше принципу их структурного единства: любая из таких молекул представима двудольным графом. Попробуем еще более контрастно взглянуть на структурные особенности этих систем.
Заметим в этой связи, что множество всех мыслимых сильно консонатных структур можно разбить на изовалентные ряды (8, 16, 24 и т. д. электронов). Используя старинное правило Гримма ("изо-валентные группы — изоструктурны", ср. F, ОН, NH2и СН3) каждый изовалентный ряд можно разбить на несколько изоструктурных. (Подробнее об этом см. ранние работы автора в сборниках [2, 63]). Каждый изоструктурный ряд можно символически представить единой структурной формулой — двудольным мультиграфом (без атомов водорода и гетероатомов, но с кратными связями), объединяющим структурно родственные молекулы внутри ряда. Так, молекулы, изовалентные и изоструктурные неопентану, изобутилену, пропину и аллену, дадут четыре ряда, представленные на схеме 4.
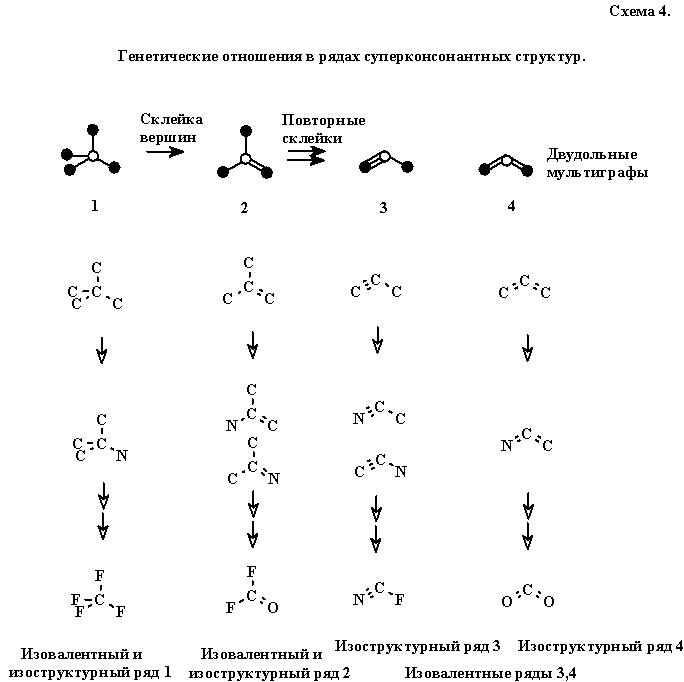
Будем считать, что изоструктурные ряды образуют один и тот же класс, если их мультиграфы имеют равное число вершин белого цвета (выражающих, как мы условились выше, электроположительный углеродныйцентр).Так, все мультиграфы 1—4 (схема 4), имея лишь один электроположительный центр (белую вершину мультиграфа) попадут в один класс. Нетрудно видеть, что мультиграфы внутри класса связаны простым генетическим отношением: структуры 2, 3 и 4 с кратными ребрами можно рассматривать как более простые производные графа 1, полученные из него путем последовательного склеивания (или слияния) между собой вершин черного цвета с обязательным сохранением числа ребер.
Нетрудно доказать, что и в общем случае сколь угодно сложный (полициклический, разветвленный) двудольный мультиграф можно получить путем аналогичной последовательной склейки неэквивалентных черных вершин какого-либо дерева (графа без циклов и кратных ребер). Так, любой мультиграф с двумя белыми вершинами (схема 5) можно получить за конечное число шагов, склеивая черные вершины исходного дерева. Понятно, что с любым деревом-прототипом ассоциирован строго замкнутый класс. В свою очередь, сами деревья-прототипы нетрудно перебирать вручную или генерировать с помощью компьютерных программ.
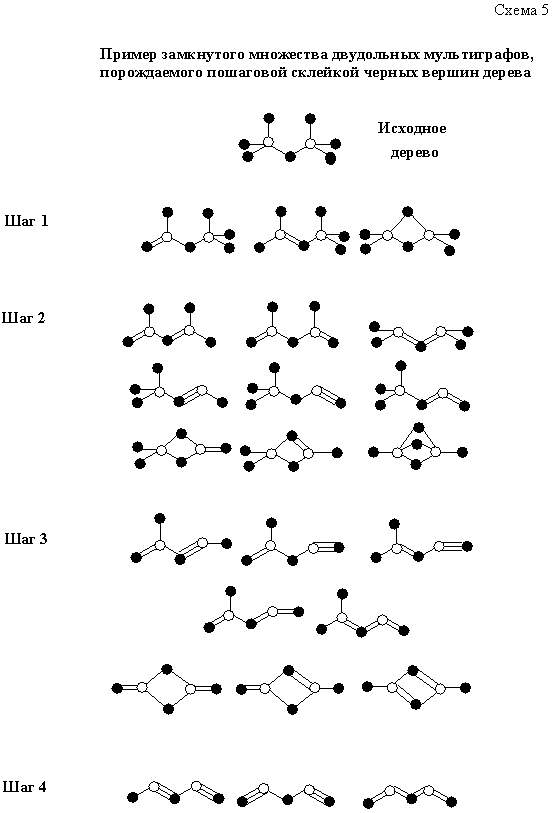
Понятно, что в дальнейшем по структуре каждого отдельно взятого мультиграфа можно восстановить обратно весь отвечающий ему изовалентный ряд (типичная задача из категории “комбинаторики электронного счета”, поставленных автором ранее [2, 63] и допускающих решение с помощью теоремы Пойа). Таким образом, структура любой (нейтральной) консонантной молекулы возникает как своеобразное совместное решение двух “уравнений симметрии”: первое — симметрия исходного дерева плюс число шагов, приводящих к нужному мультиграфу, второе — симметрия самого мультиграфа, плюс число шагов, приводящих к стандартной химической формуле.
4.4. Проблема альтернирования заряда. Развивая аналогию о сильно консонантных структурах как о “замаскированных” ковалентными связями ионных ансамблях, было бы оправданно полагать, что замеченное в квантовой химии альтернирование зарядов (см. выше) будет проявляться и для суперконсонантных молекул. При этом следует четко разграничить случаи, когда в таких молекулах ковалентные связи полярны (аналогия с ионностью оправдана) от случаев, когда связи неполярны (потенциальная ионность максимально замаскирована). Действительно, если такая молекула целиком состоит из полярных связей (неважно, простых или кратных), подобное альтернирование полных зарядов на атомах (Малликеновских заселенностей) ярко выражено. Для проявления зарядового альтернирования в неполярном фрагменте — метильной или винильной группе — требуется, как минимум, расположенная по соседству полярная связь (или несколько связей) любой кратности. Автор провел выборочное тестирование 130 таких суперконсонантных структур традиционными полуэмпирическими методами CNDO/2, MNDO, MINDO/3 и AM1 и убедился, что качественное проявление феномена для указанных случаев не зависит от выбираемого метода.Сложнее обстоит дело с неполярными структурами. В недавней работе сопоставлялись 7 альтернативных методик вычислить “заряд на атоме” в ab initio расчетах [64]. Результат оказался зависящим от методики, и для заряда углеродного атома в метане дал разброс от —0,880 до +0,244.
Полуэмпирические методы расчета зарядов в неполярных молекулах также дают противоречивые результаты. Тем не менее, анализируя данные расчетов большой выборки неполярных структур разными полуэмпирическими методами, можно прийти к любопытной закономерности. Один из методов, а именно метод AM1, систематически приписывает диполю связи С—Н полярность С(-)H(+) почти независимо от гибридизации. Иными словами, этот метод, качественно не противоречащий одному из положений рассматриваемой модели, может рассматриваться как способ тестирования модели в целом, а именно, для оценки зарядового альтернирования в углеродном скелете суперконсонантных структур углеводородов.
Результаты расчета, проведенные нами для неполярных суперконсонантных структур с простыми либо двойными связями в цепи или четном цикле, приведены на схеме 6.
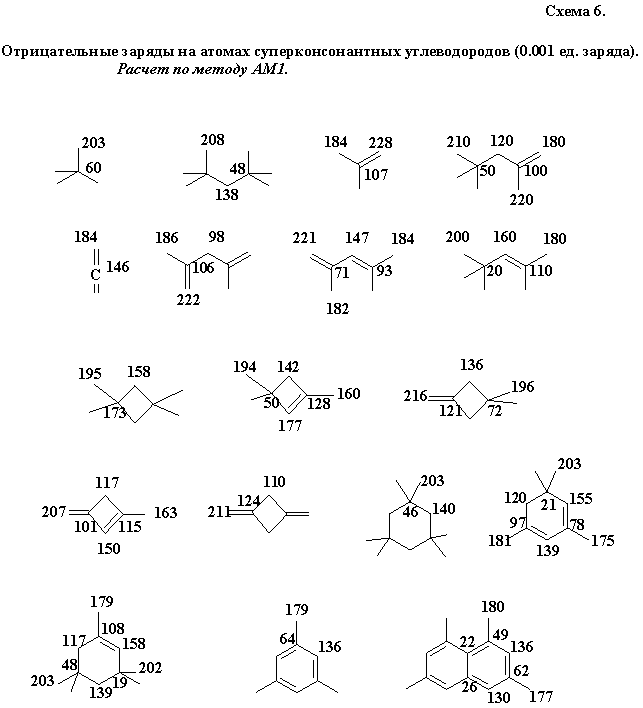
Поскольку все атомы углерода несут слабый отрицательный заряд, кажется, что альтернирования нет. На самом деле альтернирование проявляется весьма специфично, а именно, в весьма отчетливом чередовании сильного и слабого заряда углеродных атомов. В подавляющем большинстве случаев больший минус оказывается именно там, где можно локализовать гипотетический анион С4-, превосходя по величине примерно вдвое слабый заряд тех положений, где должен стоять ион С4+. (Исключения мало выраженны и касаются несопряженных двойных связей и узлового атома в нафталиновом ядре.)
Неудивительно, что это правило не выполняется для тройной связи (скажем, в случае пропина). Сама попытка связать скрытую катионоидность с sp-гибридным атомом имеет внутреннее противоречие: в тройной связи оба атома имеют наивысший s-xaрактер, а следовательно, потенциальный анионоидный тип. Тем не менее, существует способ показать скрытый электрофильный характер sp-гибридного центрального атома пропина, именно не выходя за рамки альтернирующих зарядов полуэмпирических моделях. На схеме 7 сопоставлены рассчитанные тем же методом заряды для суперконсонантных углеводородов и сопряженных им (суперконсонантных по определению) катионов и анионов, полученных депротонированием sр3-гибридного центра.
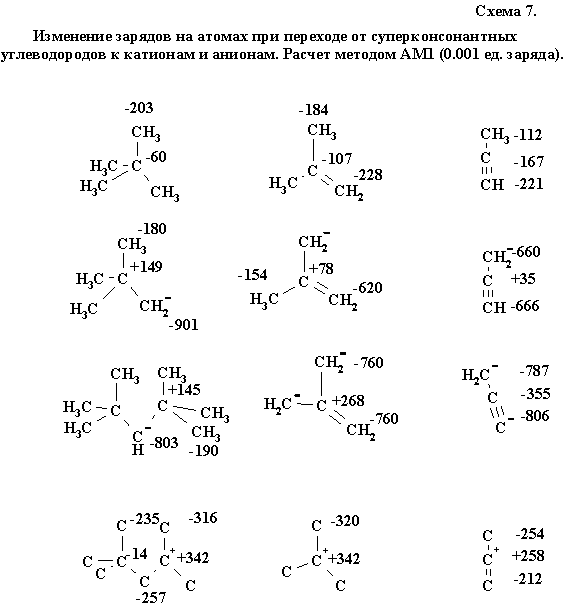
Нетрудно видеть, что как в сигма-, так и в пи-системах невыраженное альтернирование нейтрального углеводорода трансформируется в мощный градиент плюса и минуса, наблюдаемый в ионе, охватывая, в частности, и случай тройной связи пропина.
5.1. Предварительные замечания. Продолжая аналогию между суперконсонантными
структурами и ассоциатами ионов, зададимся вопросом: какие особенности
химических свойств ионных соединений можно глобально ассоциировать с химическим
поведением суперконсонантных структур? По-видимому, главное свойство иона
(отличающее его от не-иона) это его заряд. Ясно,
что относительно процессов диссоциации (растворения соли в воде), ассоциации
(формирования кристалла) или ионного обмена свойство иона нести определенный
заряд не исчезает. Уничтожить это свойство — значит отнять или добавить
электрон, скажем, путем электролиза или действия внешнего окислителя или
восстановителя.
Это совершенно тривиальное умозаключение, однако, по отношению к ковалентным суперконсонантным структурам несет принципиально иной смысловой оттенок: а обязаны ли, собственно формальные ионы, сколлапсировавшие в ковалентную суперконсонантную структуру, сохранять хотя бы "память" о своем заряде при аналогичных процессах ассоциации (присоединения), диссоциации (элиминирования) и обмена (замещения) в отсутствии внешних окислителей и восстановителей? Является ли приписываемое таким структурам топологическое свойство двудольности, где цвет жестко привязан с заряду, универсальным и неизменным инвариантом в отсутствии ред-окс процессов? Частичный ответ на вопрос (пример того, что такое иногда случается) дали Эванс и Зеебах, заметившие, что консонантность можно создать из консонантности, а нормальную цепь — из другой нормальной цепи. Оба автора, правда, не заметили, что дали конструктивный пример правила сохранения, а именно, сохранения альтернирования.
В модели Зеебаха исходно существуют нормальные структуры (или их синтоны) и некие “внешние” реагенты, полярная структура которых не сопоставляется с исходными. В нашей модели полярная структура любого реагента принципиальна: она либо суперконсонантна, либо нет. Возникает вопрос: имеются ли среди реагентов, упоминавшихся Зеебахом для создания умполунга (см. перечисленные выше 6 типов) примеры суперконсонантных частиц? Совершенно очевидно, что использование для этой цели типично карбеноидных, секстетных или септетных частиц, циклопропанов, цианид- и ацетиленид ионов — это использование некоторых внешних (по отношению к самому суперконсонантному множеству) структур, которые непредставимы в виде ассоциата "правильных" ионов, а следовательно выступают в качестве (явных или скрытых) окислителей и восстановителей. По той же причине следует отбросить ред-окс реакции. Таким образом, остается только умполунг путем замены гетероатома, который, как явствует из всех примеров, обсуждаемых в обзоре [52], не включает ни одного случая суперконсонантных структур. Таким образом, мы приходим к предварительному и не совсем тривиальному заключению, что создать умполунг (структурную диссонантность, полярную гетерогенность) реакциями только внутри множества суперконсонантных структур невозможно.
5.2. Формулировка гипотезы. Заметим, что до сих пор умполунг понимался именно как процесс создания структурного фрагмента, противоречащего нормальному альтернированию в цепи. Поскольку наше определение альтернирования с самого начала было несколько шире (включая правильное взаиморасположение водородных атомов, а также водорода и гетероатомов), постольку и умполунг (появление диссонантности) мы вынуждены трактовать также более широко, а именно, как потерю свойства суперконсонантности. В этой связи правомерно поставить вопрос: а можно ли путем только полярных реакций и только внутри суперконсонантного множества выйти за его пределы? Если бы нам удалось ответить на этот вопрос категорически отрицательно, то из этого вытекал бы фундаментальный принцип, который можно выразить в трех равновесных формулировках:(а) Сохранение ритма. Свойство суперконсонантности молекул в ходе их полярных реакций между собой имеет тенденцию к сохранению.
(б) Математическая замкнутость. Множество полярных суперконсонантных молекул стремится остаться замкнутым относительно полярных реакций, протекающих внутри этого множества.
(в) Сохранение локального свойства. Изначальная электро- или нуклеофильность атомов в суперконсонантных структурах имеет тенденцию сохраняться в ходе полярных реакций с другими такими же структурами.
5.3. Особенности реакционной способности суперконсонантных структур. Примем сформулированный принцип в качестве рабочей гипотезы, с которой-можно сопоставлять факты. Если подходить к проблеме педантично, то за более чем полтора столетия химики не так часто проводили “чистые” эксперименты между структурами строго суперконсонантными. (Традиционные кислоты и основания, третичные амины, сложные эфиры, многие стандартные растворители таковыми, очевидно, не являются.) Тем не менее, как мы уже упоминали в разделе 4.2, сильно консонантные структуры слишком хорошо знакомы химикам-органикам, и особенности их синтеза и реакционной способности давно вошли в классические учебники.
Необходимых фактов вполне достаточно, чтобы убедиться в справедливости гипотезы по крайней мере для широкого класса кислород-, азот- и фтор-содержащих структур. В большинстве своем такие процессы действительно типичные “реакции из учебника”, открытые еще в прошлом веке или начале нынешнего столетия. Достаточно припомнить, например, синтез Веллером мочевины, самоконденсацию ацетона, реакции Риттера, Реформатского, отдельные примеры реакции Михаэля, Кляйзена, перегруппировки Фаворского, различные синтезы на основе производных малоновой или ацетоуксусной кислот, классические именные синтезы пиридинов, пиримидинов, солей пириллия, пиронов и т. д.
Приходится с долей сожаления отбрасывать положительные примеры реакционной способности целых классов органических соединений (сложных эфиров, кеталей, виниловых эфиров, производных алифатических спиртов и аминов, хлор-, бром-, кремний-, бор- и алюминииорганических структур), которые явно свидетельствуют в пользу сохранения консонантности в полярных процессах, но структура которых, однако не подпадает в узкие рамки суперконсонантности.
Если считать, что определенный итог развития органической химии первой половины столетия подведен в классической многотомной серии [65], то оказывается, что до 1952 г. в химии полярных ациклических структур попросту не существовало достойного контрпримера сформулированному выше принципу. (По крайней мере, они не отражены в первых двух томах серии, содержащих бесконечное перечисление конкретных реакций алифатических соединений).
Заметим, что в истории химии есть пример последовательного использования принципа суперконсонантности в практическом дизайне карбо- и гетероциклов. Речь идет о знаменитой ацетатной гипотезе Колли [66], согласно которой ряд ароматических и гетероароматических структур можно составить из фрагментов уксусной кислоты. Известно, что именно эта модель легла в основу современных представлений о биосинтезе фенолов [67].
Вспомним, что именно замещенные бензолы выступали ранее в качестве объектов горячих дискуссий между сторонниками и противниками модели альтернирования (Так, мета-нитрование анилина в кислотах или случаи нестандартных ориентации использовались для опровержения модели). Между тем, в случае суперконсонантных бензолов (например, 1,3,5-замещенных донорными группами) практически отсутствует возможность протекания электрофильного мета-замещения по отношению к донорной группе (это потребовало бы ипсо-атаки). Напротив, именно для этих систем зарегистрированы наиболее стабильные аренониевые ионы (очевидно, также суперконсонантные), возникающие практически при любом сочетании F-, OH-, СН3- и даже NН2-групп. Вместе с тем, нуклеофильное замещение таких молекул, в отличие от других бензолов, не требует дополнительной активации (ср., например, взаимные превращения флороглюцина в 1,3,5-триаминобензол и обратно). Такие структуры, кстати, наиболее стабильны термодинамически и нередко являются конечными продуктами при изомеризации как в кислой, так и нуклеофильной средах.
Можно дать без ссылок некоторые другие известные и наиболее яркие особенности химического поведения отдельных классов сильно консонантных систем. Так, в химии алифатических фторпроизводных известно положение о том, что фторид-ион “зеркален” протону (ср. поведение изобутилена и пер-фторизобутилена под действием HF). Нетрудно вспомнить примеры, в которых циано-группа будет “зеркальна” фтору (ср. кислотные свойства гексациан-изобутилена или цианирующие свойства тетрацианметана по отношению к фторид-иону).
Хорошо известно большое число обратимых процессов и реакций расщепления суперконсонантных структур на более простые субъединицы с ожидаемым сохранением альтернирования в цепи (ср. необычно легкое расщепление бета-дикарбонильных соединений, бета-кето- и бета-дикарбоновых кислот и поликислот). Легкое раскрытие цикла претерпевают пироны и соли пириллия, а трансформацией солей 2,4,6-пириллия или коллидиния можно создать бензольное ядро. Экзотическим, но вполне тривиальным в данном контексте, является щелочное расщепление флороглюцина на ацетон, уксусную кислоту и СО2. Легкое раскрытие суперконсонантных структур с четырехчленным циклом наблюдается в случае бета-лактонов, димерных изоцианатов, при фрагментации циклобутенонов по Гробу.
Таким образом, существует большое число полярных суперконсонантных структур и полярных процессов, в которых топологическое свойство полярной двудольности, а следовательно, и полярная природа центров сохраняются. Большинство таких реакций являются ионными процессами электро- или нуклеофильного присоединения или замещения, протекающими в полярных условиях, включающих кислотный или основный катализ. Нередко это обратимые реакции, подчиняющиеся зарядовому контролю, для которых применим принцип микроскопической обратимости. Найденные нами исключения весьма немногочисленны (см. раздел 5.5).
Значительное число данных свидетельствует о том, что свойство суперконсонантности, как ни странно, сохраняется и для неполярных систем в полярных процессах. Самый банальный пример такой реакции — классические опыты Бутлерова по действию серной кислоты на изобутилен. (Ясно, что поли-изобутилен или любой олигомер изобутилена, полученный по типу “голова к хвосту”, суперконсонантны.) Другие примеры можно условно разбить на три больших класса.
Во-первых, это катализируемые кислотами или основаниями прототропные перегруппировки, не меняющие исходного альтернирования, известные для целого ряда чисто углеводородных структур (ацетилен-аллен-диеновые перегруппировки, миграция тройной связи в диинах и енинах, превращение экзо-двойной связи в эндо-связь в 1,3-диметиленциклобутане и т. д.).
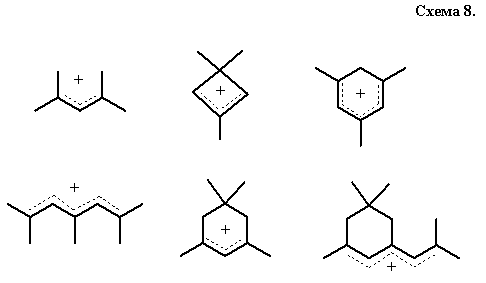
Во-вторых, это процессы протонирования и депротонирования структур с суперконсонантным расположением функций, в точности соответствующим требованию максимальной делокализации образующегося заряда. Так, хорошо известные [68] структуры наиболее устойчивых ациклических и четно-циклических карбокатионов суперконсонантны, схема 8 (ср. с данными расчетана схеме 6), причем заряд можно локализовать именно в тех позициях, где располагался бы гипотетический центр С4+.
Вместе с тем, хорошо известны стабильные моно-и полизарядные анионы суперконсонантных углеводородов с сопряженными и кросс-сопряженными связями (моно- и дианион мезитилена, дианион изобутилена, дважды и четырежды депротонированные анионы пропина и пентадиина, см. например [43]), структура и реакционная способность которых полностью согласуется с рассматриваемым принципом. Любопытным фактом газофазной химии карбанионов, генерируемых из анионов алкилкарбоновых кислот [69], оказывается неожиданная стабильность именно метильного и неопентильного анионов (являющихся суперконсонантными) в сравнении с промежуточными гомологами.
Многочисленная третья группа реакций включает появление полярной связи в исходно неполярном субстрате за счет реакции с полярными реагентами. Классические примеры — Марковниковское присоединение к изобутилену и его димерам, гидратация пропина и аллена, последовательное ацетилирование изобутилена до оксида мезитила или катиона пириллия, многочисленные реакции депротонированных ацетиленов (например, пропина) с электрофильными кратными связями.
5.4. Ретросинтез, синтетическая эквивалентность и двудольность. Широкое соблюдение простого правила в ряду полярных молекул, структура которых удовлетворяет жесткому детерминизму двудольных графов, позволяет по новому взглянуть на проблему планирования синтеза суперконсонантных структур. Если ограничиться только “логичными” (по Кори) расчленениями связей целевой структуры, то предшественники естественно обязаны быть суперконсонантными. Получается, что в рассматриваемом ряду задача ретросинтеза (предсказать все возможные предшественники и выбрать наиболее оптимальные) и задача прямого планирования результата синтеза (предсказать все возможные продукты реакции и оценить наиболее вероятные) в какой-то мере становятся симметричными, хотя бы в силу предполагаемой замкнутости множества. По крайней мере именно в этом вопросе допустимо предельное сближение позиций абстрактного математика и химика-экспериментатора, совмещение эмпирических знаний о природе вещей с мощью алгебры и комбинаторного анализа.
Мы ограничимся рассмотрением одного принципиального вопроса: какую новизну чисто математическая модель может внести в понимание синтетической эквивалентности. Хорошо известно, что некоторые сильно консонантные структуры легко превращаемы друг в друга или выступают в реакциях в качестве синтетических эквивалентов, что весьма важно при планировании синтеза более сложной углеродной цепи или цикла. (Так, “недокись” углерода заменяет малоновую кислоту или ее хлорангидрид, ацетоуксусная кислота эквивалентна дикетену и может обратимо трансформироваться в енамин, бета-галогенвинилкротоновую, бета,бета-дигалогенмасляную или тетроловую кислоты.) Нетрудно убедиться, что главным фактором, определяющим эквивалентность функций в суперконсонантном ряду, оказывается именно идентичность углеродной цепи: природа гетероатома, положение кратной связи или атомов водорода оказываются зачастую непринципиальны. Для самого простого случая такую эквивалентность можно условно выразить схемой 9, символически объединяющей взаимозаменяемые структуры в горизонтальных рядах. При этом можно вполне снять жесткое требование суперконсонантности синтонов, заменяя с известной осторожностью гетероатом X в структурах схемы 9 на подходящий заместитель (уходящую группу).
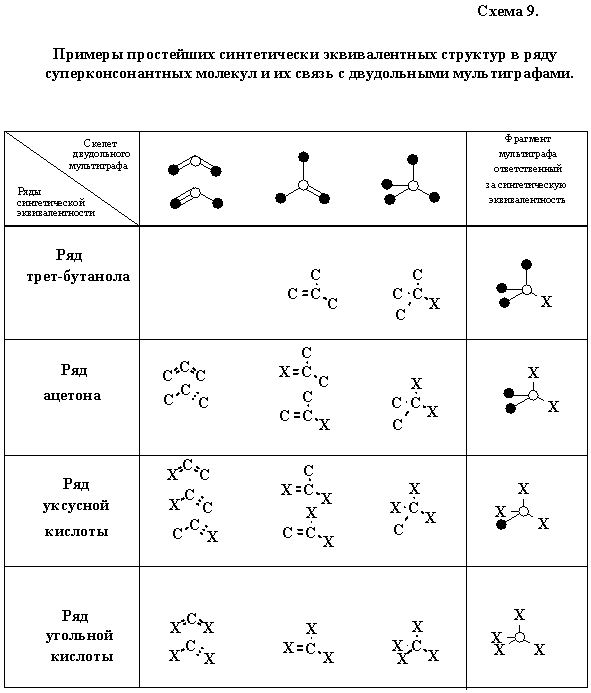
Выше, в разделе 4.3 мы показали существование простого генетического отношения между сильно консонантными структурами. Сопоставляя схему 9 со схемами 4 и 5, нетрудно видеть, что синтетически эквивалентные ряды с такой же легкостью можно описывать (и генерировать на компьютере) с использованием той же комбинаторной модели. Главное, чтобы в процессе такой склейки число атомов углерода, ответственное за синтетическую эквивалентность, оставалось неизменным.
Если синтоны схемы 9 (выводимые из схемы 4) почти тривиальны, то синтоны, которые можно получить из мультиграфов, скажем, схемы 5 и более сложных рядов, будут уже не совсем традиционны. Неожиданными эквивалентами обычных соединении могут оказаться малополярные ненасыщенные фрагменты или четные циклы. В особенности важной такая задача становится при анализе нетривиальных циклических предшественников для заданных циклических структур, в частности, для дизайна рециклизаций гетероциклов [62]. Другим возможным приложением подхода может служить использование нетривиальных синтонов в конструировании четно-циклических гетероароматических систем [58—60].
5.5. Правило и исключения. Естественно ожидать, что чем менее полярна структура, тем больше вероятность отклонения ее химического поведения от свойства сохранять суперконсонантность (трудно потерять то, чего нет). Аналогично, можно ожидать нарушений правила в случае неполярных реакций. Тем удивительнее факты сохранения суперконсонантности, нередко протекающего при термолизе. Помимо “деполимеризации” ди- и триизобутилена, можно вспомнить, например, распад 1,1,3,3-тетраметилциклобутана [70] на две молекулы изобутилена (побочно образуется также один из диизобутиленов). Разложение тринеопентилалюминия на триметилалюминий и изобутилен [71] (т. е. фактически, “гетеролиз" неопентил-аниона на суперконсонантные части) также имеет неполярный аналог в термическом расщеплении неопентана на метан и изобутилен.
Другим фактором, способным содействовать нарушению полярной двудольности, является отмечавшееся выше противоречие между потенциальной катионоидностью и sp-гибридизацией в ацетиленах и алленах. Действительно, характерной чертой химии алленов являются термические димеризации типа “голова к голове”, приводящие к четной цепи между атомами водорода или между гетероатомами (реакция типична как для аллена, так и для перфтор-аллена). Заметим, что этот факт не исключает возможности заставить ту же реакцию протекать “правильным” образом (ср. например ионную димеризацию аллена по типу “голова к хвосту” в кислой среде с образованием суперконсонантного 1,3-дигалоген-1,3-диметилциклобутана [72]. Нельзя пренебречь и отчетливой селективностью, наблюдаемой в реакциях нуклеофилов с алленами, замещенными акцепторными группами [73]. Примером прямого эффекта альтернирования в кумуленовой системе являются “зеркальные” реакции перфтораллена и пер(трифторметил)аллена: нуклеофилы реагируют с первым по терминальному атому, а со вторым — по центральному атому углерода.
В химии полярных реакций малополярных алкинов, енинов и диинов вопросов, пожалуй, больше, чем ответов [73—75]. Проблема состоит, главным образом, в несогласуемости и непредсказуемости результатов реакций гидратации и присоединения спиртов или галоидводородов для структур этого класса вообще, а в частном случае и для суперконсонантных структур. Например, пентадиин-1,3 гидратируется в кислоте с сохранением суперконсонантного мотива, тогда как спирты нормального строения (пусть не суперконстантные) атакуют концевой sp-гибридный атом [76]. Например, метилизопропенил-ацетилен, присоединяя галоидводород (по двойной связи) или алкоголят (по тройной связи) консонантным образом, подвергается гидратации по атому С-3 с нарушением суперконсонантности [75, 77]. По-видимому, на направление реакции ацетиленов с нуклеофилами могут драматически влиять стерические факторы. Так, в реакции пропина с анионом трет-бутилата появляются заметные количества анти-Марковниковского аддукта [78]. Представляется, что именно в этой области возможны примеры новых исключений.
Слабая полярность СН-связей углеводородов в подходящих условиях может быть подвергнута “неправильному” гетеролизу на гидридный и карбокатионный центр под действием мощных кислот Льюиса. Так, скелетная изомеризация неопентана под действием галогенидов алюминия вызывается, очевидно, отрывом гидрид-иона и образованием диссонантного неопентильного катиона. Известно, что протонирование метана в суперкислых средах приводит к генерации диссонантного метильного катиона, участвующего в дальнейших превращениях; трет-бутанол в 96% серной кислоте образует, вероятно по тем же причинам смесь углеводородов.
Наконец, электроциклические процессы, слабо чувствительные к полярным факторам, не обязаны подчиняться правилам региоселективности, характерным для ионных интермедиатов. Примером конкуренции между перициклической реакцией и стабилизацией суперконсонатной частицы является поведение катиона, полученного из 2,4,6-триметилгептатриена-1,3,5. Спектр катиона удается зарегистрировать, а затем структура подвергается конротаторной циклизации с замыканием диссонантного 5-членного цикла [68].
Таким образом, обсуждаемое суперконсонантное множество получается “почти замкнутым”. Химик может возразить математику, что в органической химии дважды два “почти всегда” равно четырем, хотя иногда и пяти (если не пренебрегать неполярными субстратами). Вместе с тем, случаи спонтанного нарушения полярной двудольности наиболее любопытны, как примеры истинного — а не привнесенного извне — умполунга.
5.6. Проблема новых гетероатомов. Вводя определение сильно консонантных структур, мы ограничились минимальным набором атомов и ионов. В принципе, модель можно расширить, пополнив коллекцию исходных ионов другими ионами, имеющими замкнутую электронную оболочку инертного газа. Ничто не мешает формальному описанию способов синтеза и реакционной способности таких “гомогенных”, но гетероатомных или элементоорганических систем с использованием соответствующих предельных ионных структур. Простейший пример — нитрометан, в котором легко условно вычленить ион N5+ (изоэлектронный иону С4+) или тиомочевина, “содержащая” сульфид-ион. Между тем, химия гомогенных соединений уже с этими двумя ионами будет содержать рекордное число фактов спонтанного нарушения двудольности или умполунга (ср. [47, 52]).
Представляется разумным разбить все новые ионы условно на два класса. В первый класс (сильно консонантных ионов) к 6 уже имеющимся ионам можно добавить катионы щелочных и щелочноземельных металлов, ионы В3+, Al3+, Si4+ (и возможно их аналоги), а также хлорид- и (с известной осторожностью) бромид-ион.
Второй класс будет включать оставшиеся октетные ионы, в частности, катионы N5+, S6+, P5+ и анионы S2-, B5-, I-, которые можно условно назвать слабо консонантными.
Принципиальная разница между этими классами состоит в том, что консонантные структуры, полученные комбинацией ионов первого класса, в большинстве случаев будут иметь тенденцию сохранять свойство полярной двудольности. Присутствие в молекулах ионов второго класса будет приводить как к сохранению, так и к частому нарушению этого правила. Не обсуждая детально причин этого явления, заметим отчетливую параллель с принципом ЖМКО и подчеркнем, что в самой структуре ионов второго класса имеется внутренний конфликт (хотя бы между электроотрицательностью атома, наличием вакантной орбитали и знаком заряда). В сложных органических структурах с ковалентными связями этот замаскированный конфликт будет приводить к аномальной ориентации в полярных процессах, одновременному проявлению атомом электро- или нуклеофильности, т. е. к появлению диссонантности или умполунга.
Настоящая работа щедро финансировалась грантом международного научного фонда (ISF Grant М1Х000) и грантом Санкт-Петербургского Центра фундаментального естествознания. Автор признателен докторам М. Saltzman (США) и J. Sander. (ФРГ), любезно предоставившим копии малодоступных публи-каций [8, 50] и рукописи [47].
ЛИТЕРАТУРА
1. И.Харгитаи, М.Харгитаи. Симметрия глазами химика. М., Мир, 1989, 494 с.
2. Принципы симметрии и системности в химии. Сборник статей. Ред. Н.Ф.Степанов. М., МГУ, 1987, 123 с.
3. Химические приложения теории графов и топологии. Ред. Р.Кинг. М., Мир, 1987, 560 с.
4. Concepts and Applications of Molecular Similarity. Eds. M.A.Johnson, G.A.Maggiora. J.Wiley, NY, 1990, 393 p.
5. Е.В.Бабаев, Химия и жизнь. N 4, с.17 (1989).
6. Р.Вудворд, Р.Хофман. Сохранение орбитальной симметрии. М., Мир, 1972, 206 с.
7. E.V.Babaev, Chapter 7 in: Graph Theoretical Approach to Chemical Reactivity (Understanding Chemical Reactivity, Vol.9). Eds. D.Bonchev, O.Mekenyan. Kluwer Academic Publ., Dordrecht-Boston-London, 1994, p.209.
8. A.Lapworth, Manchester Memoirs Lit. Phil. Soc., vol.64, N 3, p.1 (1920).
9. W.O.Kermack, R.Robinson. J.Chem.Soc., p.427 (1922).
10. C.K.Ingold. J.Chem.Soc. p.513 (1925); ibid pp.870, 1870.
11. A.Lapworth. Chem. Ind. (London), p.563 (1925).
12. Chem. Ind. (London), p.1050 (1925).
13. J.Shorter. Nat.Prod.Reports, vol.4, p.61 (1987).
14. C.A.Russel, ibid., p.47.; M.D.Saltzman, ibid., p.53, W.Cocker, ibid., p.68.
15. M.Saltzman. J.Chem.Educ.,Vol.49, N 11, p.750 (1972).
16. C.B.Flurcheim. J.Prakt.Chem., Bd.76, S.185 (1907).
17. D.Vorlander. Ber., Bd.52, S.263 (1919).
18. J.Steiglitz. J.Amer.Chem.Soc., Vol.44, p.1293 (1922).
19. K.Fajans. Chem.Eng.News, vol.27, N 13, p.900 (1949).
20. В.Хюккель. Теоретические основы органической химии. М., ИЛ, 1958. Т.2, сс.523, 535.
21. E.Muller. Neuere Anshanungen der Organischen Chemie, Springer, Bln., 1940, S.66.
22. A.E.Remick. Electronic Interpretation of Organic Chemistry. J.Wiley, NY, 1947, p.27.
23. J.B.Conant, W.R.Kirner. J.Amer.Chem.Soc., Vol.46, p.232 (1924).
24. J.B.Conant, R.E.Hussey. J.Amer.Chem.Soc., Vol.47, p.488 (1925).
25. E.L.Eliel in Steric Effects in Organic Chemistry. Ed. M.S.Newman. J.Wiley, NY, 1956, p.138.
26. A.Iliceto, A.Fava, A.Simeone. Gazz.Chim.Ital., Vol.90, N 4, p.660 (1960).
27. K.L.Williamson, S.Mosser, D.E.Stedman. J.Amer.Chem.Soc., Vol.93, p.7208 (1971).
28. I.Morishima, K.Yoshikawa, K.Okada, T.Yonesawa, K.Goto. J.Amer.Chem.Soc., Vol.95, p.165 (1973).
29. D.W.Davis, M.S.Banna, D.A.Shirley. J.Chem.Phys., Vol.60, p.237 (1974).
30. E.E.Ernstbrunner, J.Hudec. J.Amer.Chem.Soc., Vol.96, p.7106 (1974).
31. J.E.Lambert, D.A.Netzel, H.Sun, K.K.Lilianstrom. J.Amer.Chem.Soc., Vol.98, p.3778 (1976).
32. Л.П.Залукаев, Р.П.Воробьева, Ж.В.Шмырева, Т.А.Олейникова. Ж.Общ.Хим., т.44, N 5, с.1141 (1974).
33. S.Oae, C.A.Vander Werf. J.Amer.Chem.Soc., Vol.75, p.5037 (1953).
34. R.D.Stolow, P.W.Samal, T.W.Giants. J.Amer.Chem.Soc., Vol.103, p.197 (1981).
35. И.А.Леенсон. Чет или нечет? М., Химия, 1988, 174 с.
36. А.А.Петров. Химия алканов. М., Наука, 1974, 244 с.
37. М.Дьюар, Р.Догерти. Теория возмущений молекулярных орбиталей в органической химии. М., Мир, 1977, 695 с.
38. J.A.Pople, M.S.Gordon. J.Amer.Chem.Soc., Vol.89, p.4253 (1967).
39. W.J.Hehre, J.A.Pople. J.Amer.Chem.Soc., Vol.92, p.2191 (1970).
40. P.Politzer, S.L.Whittenburg, T.Warnheim. J.Phys.Chem., Vol.86, p.2609 (1982).
41. G.Van Hooydonk. J.Mol.Struct. (THEOCHEM), Vol.121, p.45 (1985).
42. S.W.Benson. Angew.Chem.Int.Ed.Engl., vol.17, p.812 (1978).
43. J.Klein. Tetrahedron, Vol.44, N 2, p.503 (1988).
44. T.-L. Ho. Reviews of Chem. Intermediates. Vol.9, p.117 (1988).
45. T.-L. Ho. Polarity Control for Synthesis. J.Wiley, Chichester, 1991, 403 p.
46. E.J.Corey. Pure Appl. Chem. Vol.14, p.19 (1967).
47. D.A.Evans. UCLA Physical Organic Chemistry Seminar, May 6, 1971. (Manuscript 242A: D.A.Evans, An Organizational Format for the Classification of Functional Groups. Application to the Construction of Difunctional Relationships. 25 p.)
48. I.Ugi, P.Gillespie. Angew.Chem., Bd.83, N 23, S.985.
49. R.Doenges, B.-T.Groebel, H.Nickelsen, J.Sander. J.Chem.Inf.Comput.Sci., Vol.25, N 4, p.425 (1985).
50. F.Serratosa. Buttleti Societat Catalana Cienc. Fis., Quim., Mat. Vol.1, N2, p.75 (1977). (Chem.Abstr. 89: 196341s.)
51. D.A.Evans, G.C.Andrews. Acc.Chem.Res., Vol.7, p.147 (1974).
52. D.Seebach. Angew.Chem.Int.Ed.Engl., vol.18, N 4, p.239 (1979).
53. M.Wagener, J.Gasteiger in: Software Developments in Chemistry. 4. Ed. J.Gasteiger. Springer-Verlag, 1990, p.265.
54. F.Serratosa. Organic Chemistry in Action. The Design of Organic Synthesis. Elsevier, Amsterdam, 1990.
55. A.McKillop, A.J.Boulton in: Comprehensive Heterocyclic Chemistry. Eds. A.R.Katritzky, C.W.Rees. Oxford, Pergamon Press, 1984. Vol.2, p.67.
56. Е.В.Бабаев, С.В.Цитовский. Вестник МГУ. Серия 2 (Химия), т.35, N 1, с.85 (1994).
58. Е.В.Бабаев. ХГС, N 7, с.937 (1993).
59. Д.Е.Лушников, Е.В.Бабаев. ХГС, N 10, с.1299 (1993).
60. Е.В.Бабаев, ХГС, N7, с.962 (1993).
61. Ф.Харари. Теория графов. М., Мир, 1973, 300 с.
62. E.V.Babaev, D.E.Lushnikov, N.S.Zefirov. J.Amer.Chem.Soc., Vol.115, p.2416 (1993).
63. Е.В.Бабаев в сб. История и методология естественных наук. Вып.35. Философские проблемы химии. Ред. А.П.Руденко. М., МГУ, 1988, с.121.
64. K.B.Wiberg, P.R.Rablen. J.Comput.Chem., Vol.14, N 12, p.1504 (1993).
65. Chemistry of Carbon Compounds. Ed. E.H.Rodd. Elsevier, Amsterdam. Vol.1a, pp.221-777 (1951); Vol.1b, pp.779-1462 (1952).
66. J.N.Collie. J.Chem.Soc., Vol.91, p.1806 (1907).
67. A.J.Birch. Science, Vol.156, p.202 (1967).
68. Современные проблемы физической органической химии. М., Мир, 1967, с.393.
69. R.R.Squires. Acc.Chem.Res., Vol.25, p.461 (1992).
70. T.A.Babcock. J.Amer.Chem.Soc., Vol.91, p.7622 (1969).
71. K.Ziegler. Angew.Chem., Bd.68, S.721 (1956).
72. K.Griesbaum. J.Amer.Chem.Soc., Vol.86, p.2301 (1964).
73. Houben-Weyl. Methoden der Organischen Chemie. Thieme, Stuttgart, 1977. Bd.5/2a.
74. The Chemistry of Carbon-Carbon Triple Bond. (The Chemistry of Functional Groups). Ed. S.Patai. J.Wiley, Chichester, 1978. Parts 1,2.
75. А.А.Петров. Усп.Химии, Т.29, N 9, с.1049 (1960).
76. Б.П.Гусев, В.Ф.Кучеров. Изв. АН СССР. Сер.хим. N 1, с.208 (1974).
77. Б.С.Купин, А.А.Петров. Ж.Общ.Хим. N 29, с.3738 (1959).
78. В.И.Лаба, А.А.Крон, С.П.Ситников, Е.Н.Прилежаева. Изв. АН СССР. Сер.хим. N 9, с.2129 (1972).