THE ALTERNATION RULE:
AN OLD HEURISTIC PRINCIPLE OR A NEW CONSERVATION LAW?
E. V. Babaev
INTRODUCTION
In relation to natural sciences, mathematics was and is an unattainable standard of accuracy, for which any science, studying natural phenomena, strives. One of the key features of mathematical thinking consists in separation of ideal sets, closed in relation to some operation of changes in the objects themselves of this set. Thus, a set of numbers, matrices, or polynomials can be examined, on which operations are determined (different types of addition, multiplication, etc.), not going beyond the limits of the initial sets. Examples of mathematical structures, determined by namely this way, can be algebra, groups, rings, fields, etc., generating a rich arsenal of mathematical subjects.Closed mathematical structures play an enormous role in natural science. A special case of such structures -groups - is formalized by the qualitative concept of the symmetry of objects, expressing in symbolic form intuitive concepts of proportionality, harmonics, and order of parts of the whole. The well-known applications of group theory in chemistry [1, 2] clearly show namely of what use can be analysis of the symmetry of wave functions or the symmetry of the location of atoms in molecules as points in space. In addition, the use of the idea of group theory in physics gives an example of a fundamentally different possibility of use of the apparatus of closed sets in natural science. Physicists have long noted that the presence in an object of certain symmetry, described by a closed group structure, is equivalent to the presence of a certain (explicit or latent) property of invariance, i.e., the law of conservation.
In the last decade chemistry has accumulated a rich arsenal of algebraic models, successfully used for the solution of problems of the "structure-property" type, used in computer synthesis or focused on determination of the degree of molecular similarity (e.g., see collections [2-4] and bibliographic review [5]). However, we note that of the known algebraic models not one example is known to us in which such a model would have led to the formulation of some new conservation principles. It can be stated that chemistry is generally extraordinarily poor in invariants and conservation laws. Thus, if the invariance of mass, charge, and energy is discarded (conservation laws, physical in their nature), then only the law of conservation of orbital symmetry [6] and the principle of invariance of the Euler characteristic remain [7].We will attempt in this paper to find an example of a closed set where it was earlier not looked for or was not noted. We will show that among structures, habitual to the glance of organic chemists, a class of molecules can be mathematically rigidly separated, possessing certain "rhythmic" similarity of electronic structure, associated with alteration of polar (donor and acceptor) centers. As an example of the operation of changes of such objects (called superconsonant below), we will examine general polar reactions, including stages of heterolytic formation and/or cleavage of bonds. The main idea of this approach is that the indicated set with certain assumptions can in fact be considered "almost closed" in relation to the indicated operation of changes. In its turn, such an assertion is equivalent to a new principle, nontrivial for organic chemistry, of retention of the topological property of polar bipartition (charge alternation) in polar processes. The formulation of the model is preceded by a review of early papers, devoted to the principle of alternation and the problem of consonance, in no way finding reflection to date in the domestic literature.
1. PRINCIPLE OF ALTERNATION: HISTORY AND MODERN STATE OF THE PROBLEM1.1. Alternation of polarities as a structural feature. In 1920 a Manchester professor, Arthur Lapworth, published (although in a relatively inaccessible journal) an excellent paper [8], stimulating very contradictory responses of both contemporaries and descendants. We will recall that the question concerned the so-called principle of alternating polarities, according to which a heteroatom, connected to (completely or partially) a conjugated hydrocarbon chain, causes alternation of positive and negative polarities of atoms along this chain. Such alternation explained numerous facts of the chemical behavior of conjugated systems (orientation upon substitution in benzenes, addition to polar multiple bonds, the effect of substituents on the CH-acidity, vinylogy, etc.). The heteroatom of both donor and acceptor groups (for example, the oxygen heteroatom of a carbonyl or nitro group or the nitrogen atom of a nitrile group, in the last case) was examined as the key atom, responsible for such alternation. The idea of Lapworth was confirmed by a series of scientists, particularly Robinson, who proposed a similar model [9].
The apparent universality combined with vagueness of formulations made the Lapworth model very vulnerable, which gave rise to a prolonged dispute between Lapworth and Robinson, on one hand, and with Ingold, on the other hand. A series of factors seemed to contradict the rule, which Ingold attempted to demonstrate in a series of papers under the name "The nature of the alternation effect in the carbon chain" [10], directed toward refuting this model. In the connection that Ingold incorrectly interpreted the initial principles of the model, and thus himself allowed a series of errors (including experimental), the discussion assumed very sharp and emotional character [11]. In 1926 the British Royal Society found a very original way out of the situation, imposing a unique veto on continuation of the dispute, calling it "games in chemical x"s and o's," and refusing "henceforth to examine and publish papers, devoted to the mysticism of polarities" [12]. Terminating the discussion, Lapworth and Robinson actually stopped the further development of the model, while the language of electronic effects, introduced by Ingold, completely displaced the terminology of alternation. Materials of science historians on this question were published relatively recently [13, 14], and attention of chemists was first drawn [15] to the name of Lapworth only in 1972 (a year after the death of Ingold).
We note that the actual ban of the further development of the idea in the British Isles did not weaken the interest in it of the world society. In addition to Lapworth, for example, similar ideas were stated by other European [16, 17] and American chemists [18, 19]. A series of pupils up to the middle of the century referred to the principle of alternation not only from a critical point of view, but also as a useful theoretical model [20-22]. It is clear that the concepts of mesomerism and alternation are deeply synonymous and are useful to an equal degree for the description of electron-density redistribution in pi-systems with heteroatoms. Nonetheless, the majority of data confirmed the model, introduced by Ingold, of damping of the effect of the polar group along the saturated chain. In addition, the attractive paradigm of alternation as a unique "idea-phantom," continued to be rediscovered anew by experimenters and episodically arose and disappeared from the visual field of theoreticians. The effect of alternation in a saturated chain, observed experimentally for individual classes and confirmed by both spectral and kinetic investigations, attracted [19, 23-26] and continues to attract special attention [27-32]. However, the expected effect was not observed in a series of cases [33, 34]. We will not dwell on known examples of alternation of physical properties [35], typical, for example, in the alkane series [36].
Understandably the idea of alternation of polarities did not remain unnoticed from the direction of quantum chemistry. Alternation of charges in alternant pi-systems with heteroatoms is even displayed in the simplest Huckel MO method. Less trivial is the appearance of a charge with the opposite sign on the central atom in the allyl cation and anion, appearing upon the use of PMO (perturbation of molecular orbitals) theory [37]. The unique renaissance of the principle of alternation in the 1970s can be associated with the appearance of a paper by Pople and Gordon [38], when the first testing of the semiempirical CNDO/2 method on a broad class of structures showed clear alternation of charge, including in saturated chains. The same result was confirmed later by ab initio calculations [39] (also see the critical analysis and bibliography in [40]). The reasons for the phenomenon are possibly more profound from a physical point of view [41].
Interesting results were obtained during the analysis of the alternation principle from the thermodynamic point of view. Thermochemical data indicate [42] that for small molecules the proximity of functions, similar in polarity, near one atom (i.e., pairs of donors of pairs of acceptors, leading to an alternating sequence) is energetically more favorable than is proximity of functions of opposite nature (i.e., with disturbance of alternation), which is reflected in the direction of disproportionation reactions. Analogously the stability of substituted olefins or benzenes is primarily determined by the alternating (or nonalternating) surrounding of the double bond (benzene nucleus) with polar groups. In particular, a result of this rule [43] is the unexpectedly higher stability of cross-conjugated fragments over systems with normal conjugation, displayed particularly clearly in the case of carbanions. Deserving attention are the recently published review and monographs of Ho, a famous American synthetic chemist [44, 45]. An attempt was made in his developed approach to use the alternation principle as an instrument to explain the most diverse facts of reactivity and regioselectivity in organic chemistry on the basis of the assumption that an alternating arrangement of functions around an atom is always more energetically favorable than upon the absence of such alternation. The discussed group of facts includes, for example, the problem of substrate activation (by both an increase and disturbance of alternation), the explanation of the observed regioselectivity in substitution, oxidation, addition, and cycloaddition processes, and the driving force of rearrangements. The author himself proposes that the principle of alternation be regarded as a convenient mnemonic rule, making it possible to solve specific problems.
1.2. Principle of alternation and design of synthesis. The principle of alternation has evidently found the greatest number of applications in the field of the design of organic synthesis. The inception is assumed to be the paper of Corey [46], in which the retrosynthesis operation was introduced, i.e., the imaginary heterolytic separation of the whole structure into arbitrary subunits of synthones. Such separation was defined as "logical" or "illogical" (logical and illogical disconnection) as a function of how much the polar structure of the formed synthone corresponded to the normal distribution of polarities in the structure of the actual reactant (comp. "logical" acylium cation and "illogical'' acyl anion). From this definition it was just a step to analysis of interrelations between alternation of polarities in the whole structure with such alternation in its precursors.
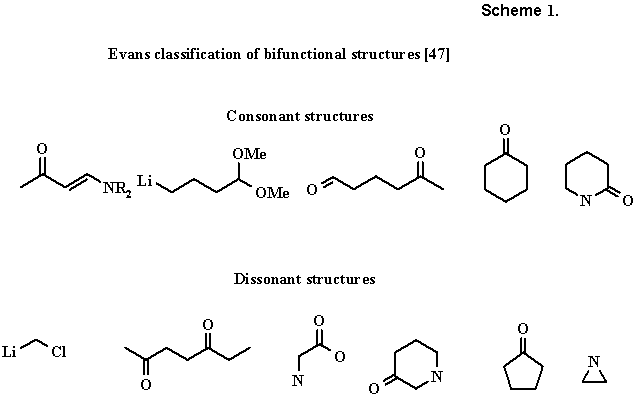
In May 1971 D. A. Evans presented a paper at a university seminar [47], which had a decisive effect on formation of a whole series of mathematical models of organic synthesis. The manuscript of Evans was occasionally circulated among many chemists, but was never published; nevertheless, this paper has repeatedly between referenced (e.g., see [48,49]), and there is detailed commentary on it in the Catalonian language [50]. In this paper Evans proposed a simple and elegant classification of bifunctional (i.e., containing two polar functions) molecules into consonant and dissonant molecules. Consonant structures permit marking of carbon atoms in such a way so that they resemble a preset pattern (charge affinity pattern) with an alternating sequence of electro- and nucleophile centers. Such a presentation is not possible in dissonant molecules (Scheme 1).
We note that the fundamental restrictions to chain saturation (as in the Lapworth model) were not imposed. Evidently Evans was one of the first to formulate strictly mathematically how namely the maximum use can be obtained of functionalities, already available in the molecule, during the design of its synthesis, having noted that the synthesis of a consonant structure (or a consonant chain of a complex molecule) is conveniently achieved from consonant precursors.It is significant that not only an electronegative heteroatom (N, O, F as in Lapworth), but also a heteroorganic fragment (with a metal of group 1 or 2, aluminum or silicon), having the opposite polarization of centers in the chain, can act as terminal groups of the bifunctional molecule or pattern with Evans. The nitro group and functions, containing sulfur, phosphorus, boron, and other elements, which led to an ambiguous distribution of polarities in the chain, separating the same atom simultaneously with electro- and nucleophiles, were separated into a special class. In experimental papers Evans [51] discussed other methods, making it possible to invert the initial consonance of the alternating chain.
The famous review of Seebach appeared slightly later [52], in which (with reference to a private communication of Evans) a more simplified and alternative terminology was derived for separation of polar structures into "normal" structures (i.e., with an alternating sequence of donor-acceptor centers in the chain) and structures, containing an "umpolung" (inversion of polarity of some donor or acceptor carbon center in relation to the heteroatom at the end of the chain). Limiting himself to an examination of only nitrogen and oxygen as the heteroatoms, Seebach superficially mentioned the synthesis of structures of normal construction from other normal precursors (reactions of the aldol or Claisen condensation type, the Prince, Mannich, and Michael reactions), considering this regularity a unique synthetic restriction.
The main merit of Seebach is the detailed analysis of methods (a series of which was first proposed by his investigative group), with the use of which an umpolung can be created. We note that a double meaning is incorporated into this term from the very beginning: an umpolung as a structural feature (e.g., parity of the chain between a pair of heteroatoms) and an umpolung as a process, in which the donor or acceptor nature of the atom is inverted. Namely which methods make it possible to create an umpolung are fundamental for our further analysis. In the opinion of Seebach, six such methods should be distinguished:(1) 1,2N-oxidation;It is understood that the initial reactants in relation to all of these methods appear as structures of normal type. The concept of a straight umpolung (e.g., in carbon monoxide, isonitriles, etc.) is formulated as a structural feature as the limiting case of an umpolung. Finally, reversal of the umpolung is regarded as a process, leading to structures of normal type.
(2) exchange and modification of the heteroatom;
(3) homologization and its reversal;
(4) the use of cyclopropanes;
(5) the use of acetylenes;
(6) redox reactions.It is not by chance that the dichotomous classification of structures and reactants into consonant and dissonant (or normal and with an umpolung), constructed on a purely mathematical definition, attracted the attention of specialists in the field of computer synthesis. The program TOSCA was developed by specialists of the German concern Hoechst AG over a period of five years [49] and led to the prediction of new ways of synthesis of artificial sweeteners with a consonant structure. A detailed formalization of the transition from consonant synthones to actual structures of reactants is used in the STRATOS program [53]. Finally, a third computer program CHAOS [54], based on analysis of ways of synthesis of bifunctional molecules, has recommended itself as a convenient medium for the study of principles of computer synthesis.
Using the alternation model for creation of computer programs, the authors introduced refinements and more clearly defined the field of applicability of the initial model. This pertains in particular to [49], where it was emphasized that the most promising model of consonant and dissonant reactants can be found in the field of heterocyclic synthesis, where polar processes (e.g., cyclocondensation) play a key role. We note that the principle of alternation for design of a heterocyclic synthesis has clearly been insufficiently recognized in heterocyclic chemistry itself. The only time the alternation rule was slipped in was recently mentioned by the authors of a review [55] during an attempt to generalize data on methods of construction of six-membered hetarenes.
Such a situation stimulated the author of the present paper to carry out a detailed analysis of the types of polar structure of reactants, ever used for the construction of the pyridine nucleus. The results of a literature analysis made it possible to create a computer data base [56, 57], the analysis of which led to the conclusion that in 95% of the cases a magic "structure-synthesis" rule [58] is observed, namely, that the alternating polarity of centers of the pyridine nucleus is rigidly determined by the polarity of acyclic reactants. Only in isolated cases were reagents with an umpolung used for the synthesis of rings (frequently with side formation of pyrroles), or a process of umpolung reversal occurred. This rule (also strictly observed for quinolines) was used as the base of the Heterocycland computer program [59], making it possible to enumerate polar types of reactants for the synthesis of hetarenes and to predict earlier unknown ways of synthesis. The experimental confirmation of the model was achieved in a new, earlier unknown synthesis of quinolines [60].
A conducted analysis
of literature on the problem of alternation clearly shows the existence
of a significant phenomenon, observed in very remote fields of both theoretical
and experimental chemistry. Unfortunately, the frequent absence of mutual
references by authors of the approaches hinders the development of a single
terminology. The authors of programs, for example, preferred to describe
the structure of molecules in terms of consonance and dissonance, using
the term umpolung namely to describe reactions. (The terminology, introduced
by Ho [45], who proposed the use of the terms "conjoint" and "disjoint"
for consonant and dissonant structures, additionally complicates the language
problem.) Since, in our opinion, the "musical-linguistic" terms consonance
and dissonance are most easily formalized, we will also follow the terminology
of Evans.
Let us now examine namely what aspects of the alternation problem are not completely elucidated and deserve further development. First, it is not completely clear if consonance should be considered a local property of the atom (e.g., due to its environment), a local property of the chain or fragment (bifunctional relation between the pair of polar functions), or a global property of the polyfunctional molecule as a whole.Second, a very serious aspect of the problem is the disregard or neglect of the degree of chain unsaturation. It was asserted beginning with Lapworth that the alternation effect is displayed most strongly in a conjugated chain; nevertheless, attempts to generalize the applicability of the effect to partially unsaturated or completely saturated chains have not ceased to date. However, in saturated chains with a polar group at the end competition between the alternation effect and the quenching effect is resolved in favor of the latter. The maximum, which can be recorded experimentally, is the saw-tooth dependence of the alternation effect, quenching at approximately the fifth unit of the saturated chain [23-32]. Analogously, during the design of the synthesis of a quite long (assume that it is consonant, but saturated) chain the selection between consonant or dissonant precursors may not be fundamental.
The third problem, virtually not touched upon on the cited approaches, can be formulated thus: is a powerful (electropositive or electronegative) noncarbon substituent in fact necessary for manifestation of alternation. Chemistry is full of examples, in which an expressed polar effect is displayed, for example, by alkyl, alkenyl, or alkyl groups. In addition, charged carbon fragments (carbocations and carbanions), i.e., synthones without heteroatoms, are the reality of modern organic synthesis. The above-cited review of Klein [43] indicates that the alternation rule has prospects of application in the field of pi-systems of low polarity.
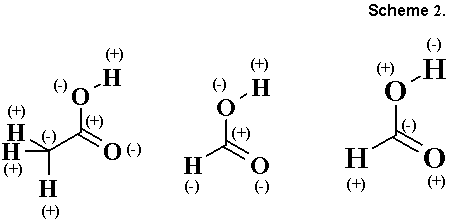
A feasible approach, which would make it possible to answer the set questions, should consider in some way the polarity of the C-H bonds. We note that this question was asked yet by Lapworth, who thought that hydrogen can be conditionally considered to be an electropositive atom, equivalent to other "key" heteroatoms. Anticipating the model of consonant and dissonant structures, Lapworth proposed to consider "homogeneous" and "heterogeneous" [8] mutual arrangement of hydrogen and heteroatoms in the chain (also see the commentary of Robinson [9] on this approach). Thus, acetic acid is a homogeneous structure, since it permits unambiguous marking of atoms by plus and minus signs, while formic acid is heterogeneous and can be marked by two methods as a function of which atom, hydrogen or oxygen, is taken as the key atom (Scheme 2). Lapworth limited himself to the possibility of introduction of such a classification and did not return to its development.
3. PRINCIPLE OF SUPERCONSONANCE
3.1. Mathematical model of polar homogeneity. Let us examine to what the joint examination of the qualitative concept "homogeneity" according to Lapworth and the stricter formulation "nonconsonance" according to Evans can lead. We will take an arbitrary organic structure, containing the N, O and F heteroatoms, in addition to C and H atoms. (Restriction to namely these elements, organogens, has a clear meaning; see concluding section.) We will assume that as normally a conditional minus sign can always be placed on the heteroatom (by virtue of its electronegativity), while we will always write a formal plus sign on the hydrogen atom; carbon atoms will be considered "neither," i.e., passive conductors of effects of "key atoms," H, N, O, and F. It is clear that polar homogeneity (the possibility to mark unambiguously with plus and minus signs carbons in the molecule) is only permissible upon strict fixation of the mutual arrangement of terminal hydrogen atoms and heteroatoms.
We will call a certain chain between the arbitrary pair of key atoms in the structure a homogeneous chain if:- the chain is odd between any pair of heteroatoms;We will consider as a special case the odd chain of a one-carbon fragment and will include its absence in determination of the even chain (i.e., direct vicinity of the function). It is understood that all carbon atoms of a homogeneous chain permit a strict unambiguous marking with alternating signs (let us say, with the same pluses and minuses).
- the chain is even between any of the heteroatoms (N, O, F) and the hydrogen atom;
- the chain is odd between any pair of hydrogen atoms.Definition 1. Molecules, constructed only from homogeneous chains, will be called homogeneous.
It is evident that a long chain can be made up of smaller chains and that a ring can also be presented as a closed chain. Consequently, rings (including connected) or structures, containing a heteroatom inside the chain (particularly heterocycles), can be included in the examination. Two ways exist of construction of sets of such homogeneous molecules. The first way is analytical: having an innumerable set of structures, we establish a filter on the basis of definition 1, separating homogeneous structures from those which are not homogeneous. The second way is constructive generation of homogeneous structures on the basis of an increase by an arbitrary (saturated or unsaturated) chain of new atoms in order that exclusively homogeneous chains (rings) are formed.
It is seen from comparison of homogeneous structures with consonant structures that these sets intersect only partially. Thus, tert-butanol, acetone, and acetic and carbonic acid are homogeneous and consonant, while methanol, formaldehyde, acetaldehyde, or formic acid are consonant, but not homogeneous. In addition, by virtue of the definition the group of homogeneous structures should include slightly polar or nonpolar structures of the propyne, isobutylene, or neopentane type, generally not described in terms of consonance.
However, the Lapworth model, obtained by refining to a logical end, leaves a certain feeling of dissatisfaction, without answers to all questions, set above in Section 2. First, the definition does not include a concept of charged species, as a result of which it is necessary to identify formally isostructural cations and anions, fundamentally differing in their polarity and reactivity (cf. an "illogical" acyl anion and a "logical" acylium cation, electrophilic OH or NH2 cations, and nucleophilic hydroxyl and amide anions). Finally, carbenoid (methylene, difluoromethylene, nitrene) and noncarbene species are in no way distinguished in the model. Thus, it is necessary that the classification include in explicit form a concept of the mutual arrangement of centers of Lewis acidity and basicity, i.e., vacancies and unshared pairs. Let us examine how the necessary results can be attained.
3.2. Generalization of the model of polar homogeneity for ions. To describe transfer of certain polar effects of hydrogen or a heteroatom (e.g. hyperconjugation) chemists long ago devised a convenient methodical procedure: imaginary heterolytic dissociation of the bond with formation of a limiting (unbonded) resonance structure of two ions. For example, we can cleave the C-H bond in the methyl group of isobutylene (into a proton and a carbanion portion) or the C-F bond in perfluorobutylene (into fluoride ion and a carbocation part) for a graphic explanation of the effect of the methyl or trifluoromethyl group. Formally nothing interferes with the repetition of this procedure many times until the covalent structure is separated into a multiply charged carbon skeleton, surrounded by a group of external ions (protons or fluoride ions in our example), see Scheme 3. In the general case this operation can also be used for heteroatoms of the oxygen and nitrogen type (generating saturated oxide- and nitride-ion structures).
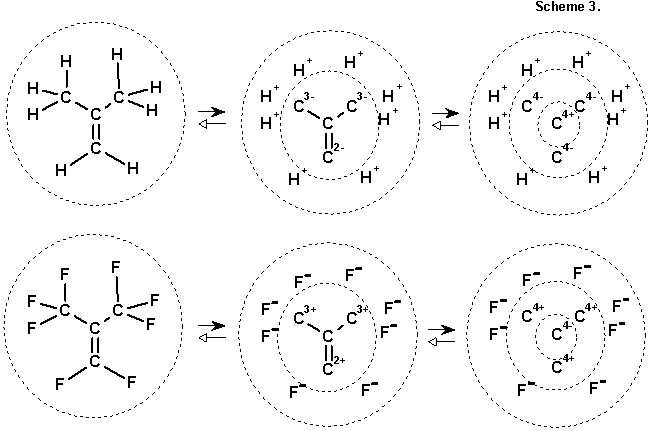
In the case of such separation of homogeneous structures the formed carbon skeleton possesses a remarkable property. According to the definition, the carbon atoms in such structures already have the plus and minus signs, and upon separation of ions the localized charge has the same sign as the initial mark. Since an alternating structure is again generated, nothing prevents the bringing of the procedure of ionic separation to a logical end by heterolytically cleaving the C-C bond with formation of limiting ions C4- and C4+ (see Scheme 3). By other words, the following remarkable theorem 1 is valid:Any homogeneous molecules can be mentally broken up into an alternating set of ions (C4+, H+) and (C4-, N3-, O2-, F-).It is evident that a nonhomogeneous molecule cannot be presented in this way - an "extra" ion will compulsorily appear, in addition to the enumerated six (cf. structures of hydrazine, ethane, or methylamine). In addition, the procedure of separation into ions can be reversed, and the sequence of assembly of a certain molecule from the indicated set of ions can be examined in such a way so that a cation and anion are always adjacent. It can be proposed that after such assembly a reorganization of the hypothetical ionic bonds will occur to the normal covalent (polar or nonpolar) bonds of the necessary multiplicity in accordance with the normal valence laws. It is not difficult to show that the following (opposite) theorem 2 will be valid in this case:Any homogeneous molecule can be assembled mentally from the supply of (C4+, H+) and (C4-, N3-, O2-, F-) ions, during which each cation is connected to an anion.Thus, polar homogeneous structures can be constructively generated not only on the basis of evenness of chains between the "key" atoms, giving the carbon atoms a passive role, but also, purely formally, on the basis of the concept of two (cationoid and anionoid) carbon atoms as equitable subunits of homogeneous structures. In addition, the implicit requirement for electroneutrality of resulting structures becomes extremely optional: one can decide on an arbitrary mono- or polycharged ion or a zwitter ion. The obtained set is already a certain qualitatively new object, for which a new name can be proposed.Definition 2. Molecules and ions, which can be mentally separated exclusively into (C4+, H+) and (C4-, N3-, O2-, F-) ions or can be assembled from these ions so that each cation is connected to an anion will be called strongly consonant or superconsonant. (We will use these same terms for the initial supply of ions).
A mathematician could say that a certain equivalence relationship is introduced by this definition into the set of molecular structures. In fact, any molecule is strictly either superconsonant (and then equivalent to other such structures) or not.
The concept of covalent molecules as a group of ions has a long tradition: namely thus in the past did Robinson attempt to visualize a picture of transfer of the substituent effect [9] and namely thus did Fijans [19], including two types of carbon ions, draw himself a picture of alternation. The problem was that this principle was given too broad an interpretation, and it was used in those cases when it possibly was not very applicable. (See [20] for the reasons for criticism of concepts of Robinson on "strengthening and disintegration of octets" along the alternating chain.) Our problem is different: having given a strict definition of a certain class, to determine the qualitative features of the electronic structure, the activity, and methods of synthesis of structures of this class.
4.1. Main features. The first purely topological feature of superconsonant structures is that any such structure is described by a bipartite graph. As is known from graph theory [61], a graph is called bipartite if the set of its apices can be broken up into two nonintersecting subsets (let us say, of different color), so that the edge of the graph connects compulsorily vertices of different color. Bipartition of a graph automatically excludes odd rings. With the use of our earlier introduced symbolism [58, 62] to assign color to charge ("philicity"), we will agree graphically to present a cationic center by a white label of a vertex in the graph and an anionoid center by a black label. We will show below that bipartition generates a very elegant genetic relation inside the class of superconsonant structures.The second features pertain to the mutual arrangement of actually charged centers, lone pairs, and heteroatoms. In the simplest cases the carbocationic center of such structures, by definition, is surrounded by atoms with lone electron pairs, and the carbanionic center is surrounded by electropositive carbon-containing fragments of the acetyl-, carboxy-, or cyano-group type (this covers cases of mesomeric charge stabilization). On the other hand, an example of inductive stabilization of the ion (carbocation - by methyl, and carbanion - by trifluoromethyl group) can also be considered a characteristic feature of superconsonant structures. Typical stabilized ions are the familiar tert-butyl cation or guanidinium, anions of typical CH-acids, perfluoro-tert-butyl and carbonate anions.
Cyanide or acetylenide ions, like the simplest methyl cation, evidently contradict the definition of superconsonance. It is also understood that the carbene or nitrene type of structure is not compatible with this definition. Finally, the ammonium or hydroxonium ion fits this definition, while tetramethylammonium or trimethylonoxinium ion does not.
The last, almost evident property, is that if the structure is superconsonant, then the sum of its valence electrons is always a multiple of eight (the reverse assertion is not true). This automatically follows from the presentation of such structures by octet anions (8 electrons are introduced) and C4+ and H+ cations without valence electrons.
4.2. "Superconsonant world." Let us examine in greater detail the structural fragments, which can be encountered among superconsonant molecules. We emphasize that because of the formal ionic analogy, arising from definition 2, any such molecule can be regarded as a unique "unsuccessful" salt. The knowledge of such structures can preferably be called recollection: too many trivial (or having trivial names) compounds fall under definition 2. It is easy to see that this set includes, for example, acetone and its oligocondensation products (mesityl oxide, phorone, isophorone, mesitylene), classical CH-acids (malonic, acetoacetic, and acetonedicarboxylic acids and their nitriles, acetylacetone, dimedone, Meldrum's acid) and their enolic forms, or enamines, ketene and its dimer, carbonic acid and carbon suboxide (C3O2).
Among the aromatic structures the example of strongly consonant structures are orcinol, phloroglucinol, symm-xylidine, and some benzoic acids like the orsellinic, gyrophoric, or lecanoric. Nitrogen-containing structures with fragments of cyanamide, urea, and guanidine are diverse. Among heterocycles can be mentioned the Guareshi imide, 2,4,6-trimethylpyrillium, symm-collidine, dehydroacetic, barbituric, and cyanuric acids, triacetonamine.It is clear that dipartition (ban of oddness of the ring) limits superconsonant structures to even-ring classes, for example, substituted b- or d-lactones, cyclobutanes and cyclohexanes, adamantanes or cubanes. With respect to types of chains of open structures, characteristic examples will be a saturated chain of polyisobutylene, a conjugated polyene chain with an alternating arrangement of a donor and a proton (or a donor and an acceptor) and also a polyine or (odd) polycumulene chain.
We note that three significant restrictions limit the full-fledged realization of the superconsonant motif. First, this is tautomerism, understood in the global sense, hindering the geminal mutual arrangement of acidic functions (cf. hypothetical orthocarbonic acid, acetone hydrate, and analogous gem-derivatives), and also the proximity of a proton-donor function with a multiple bond (hydroxy or aminoacetylene, simplest enols). Second, there is the steric effect of strongly branched superconsonant fragments of the neopentyl- or tert-butyl-group type [comp. tri(tert-butyl)amine, tetra(tert-butyl)methane, etc.]. Third, these are factors, affecting the stabilization of the pi-system (thus, the superconsonant 1,3-dimethylcyclobutadiene is antiaromatic). We note that the stabilizing aromatic effect can be consistent with the superconsonant motif (in the case of the appropriate example of substituted benzenes, naphthalenes, etc.) or can contradict it. Thus, known dianions of 1,3-dimethylcyclobutadiene or 1,3,5,7-tetramethylcyclooctatetraene are alternant and aromatic, but nevertheless, are not separated into superconsonant ions.
4.3. Genetic relations in the series of superconsonant structures. The noted structural diversity of strongly consonant molecules does not, however, contradict the above-formulated principle of their structural unity: any of such molecules is represented by a bipartite graph. Let us try to look at structural features of these systems in even greater contrast.
We note in this connection that the set of all imaginary strongly consonant structures can be divided into isovalent series (8, 16, 24, etc. electrons). With the use of the ancient rule of Grimm ("isovalent groups -isostructural," cf. F, OH, NH2 and CH3) each isovalent series can be broken up into several isostructural groups. (See early papers of the author in collections [2, 63] for greater detail.) Each isostructural series can be presented symbolically by one structural formula - a bipartite multigraph (without hydrogen atoms and heteroatoms, but with multiple bonds), connecting structurally related molecules inside series. Thus, molecules, isovalent and isostructural with neopentane, isobutylene, propyne, and allene, will give four series, presented in Scheme 4.
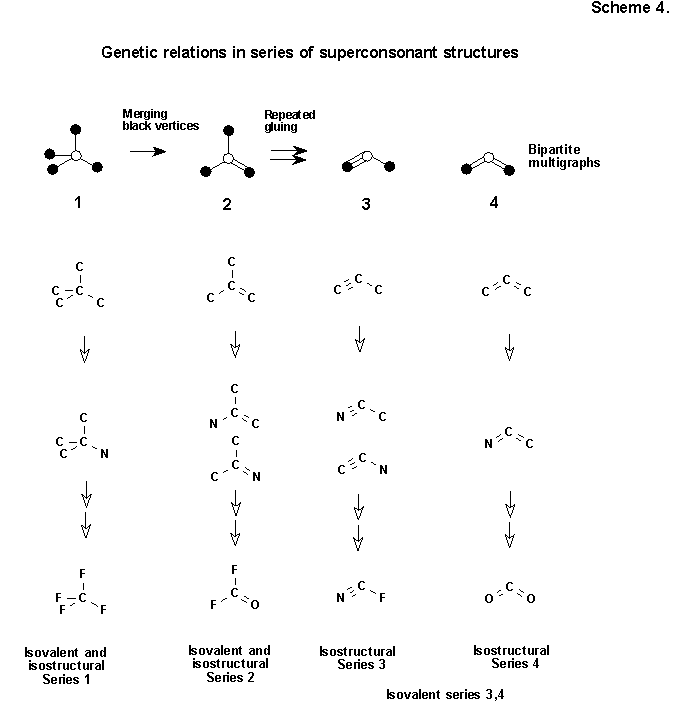
We will consider that isostructural series form one class if their multigraphs have an equal number of white apices (expressing, as we agreed above, an electropositive carbon center). Thus, all multigraphs 1-4 (Scheme 4), having only one electropositive center (white labeled vertex of the multigraph), fall into one class. It is not difficult to see that multigraphs inside a class are related by a simple genetic relation: structures 2, 3, and 4 with short edges can be regarded as simpler derivatives of graph 1, obtained from it by consecutive splicing (or merging) with each other of black vertices with compulsory retention of the number of edges.
It is not difficult to demonstrate that even in the general case as complex a dipartite multigraph as desired (polycyclic, branched) can be obtained by the analogous consecutive splicing (gluing0 of nonequivalent black vertices of some tree (graph without rings and multiple edges). Thus, any multigraph with two white apices (Scheme 5) can be obtained in a finite number of steps upon splicing black vertices of the initial tree. It is understood that a strictly closed class is associated with any prototype tree. In its turn, the prototype trees themselves are easily to enumerate manually or to generate with the use of computer programs.
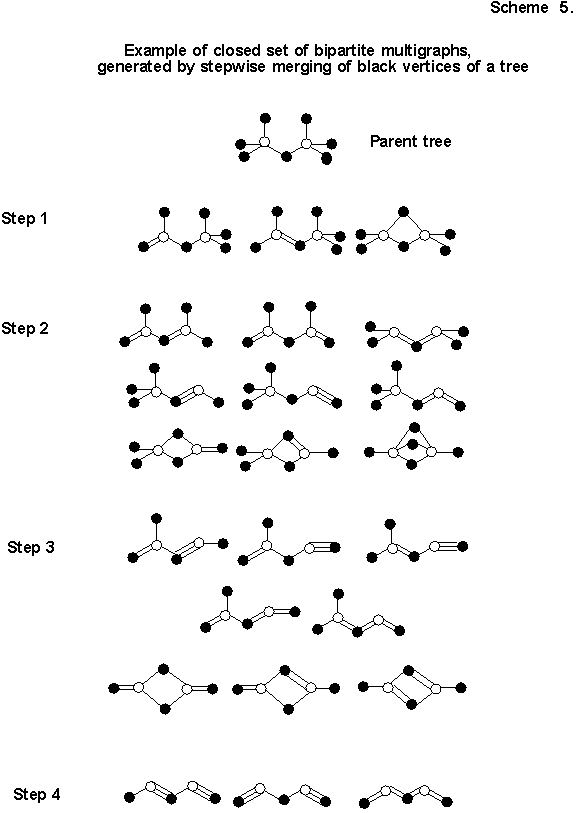
It is understandable that from the structure of any separately taken multigraph one can restore and generate the whole isovalent series, corresponding to it (a typical problem from the category of "combinatics of electron count" set earlier by the author [2, 63] and permitting a solution with the use of the Polia theorem). Thus, the structure of any (neutral) consonant molecule arises as a unique joint solution of two "symmetry equations": the first is the symmetry of the initial tree, plus the number of steps, leading to the required multigraph; the second is the symmetry of the multigraph itself, plus the number of steps, leading to the standard chemical formula.
4.4. The problem of charge alternation. Developing the analogy with strongly consonant structures as ionic ensembles "masked" with covalent bonds, we can justifiably assume that alternation of charges (see above), noted in quantum chemistry, will also be displayed for superconsonant molecules. At the same time, cases when the covalent bonds in such molecules are polar (the analogy with ionic character is justified) should clearly be distinguished from cases when the bonds are nonpolar (potential ionic character is maximally masked). In fact, if such a molecule completely consists of polar bonds (it is not important whether they are single or multiple), such alternation of total charges on atoms (Mulliken populations) is expressed clearly. For the manifestation of charge alternation in the nonpolar fragment (methyl or vinyl group) the location in the proximity of the polar bond (or several bonds) of any multiplicity is necessary as a minimum. The author carried out the random testing of 130 such superconsonant structures by the traditional CNDO/2, MNDO, MINDO/3, and AM1 semiempirical methods and determined that the qualitative manifestation of the phenomenon for the indicated cases does not depend on the selected method.
The case with nonpolar structures is more complex. In a recent paper seven alternative methods were compared for ab initio calculation of the "atomic charge" [64]. The result turned out to be dependent on the method and gave a scatter of from -0.880 to +0.244 for the charge of a carbon atom in methane.Semiempirical methods of calculation of charges in nonpolar molecules also give contradictory results. Nevertheless, analyzing data of calculations of a large sampling of nonpolar structures with different semiempirical methods, we can come to a curious regularity. One of the methods, namely the AM1 method, systematically assigns the polarity C(-)H(+) to the dipole of the C-H bond almost independently of hybridization. In other words, this method, not qualitatively contradicting any of the positions of the examined model, can be regarded as a method of testing of the model as a whole, namely, for estimation of the charge alternation in the carbon skeleton of superconsonant hydrocarbon structures.
Results of calculations, which we carried out for nonpolar superconsonant structures with single or double bonds in the chain or an even ring, are presented in
Scheme 6.
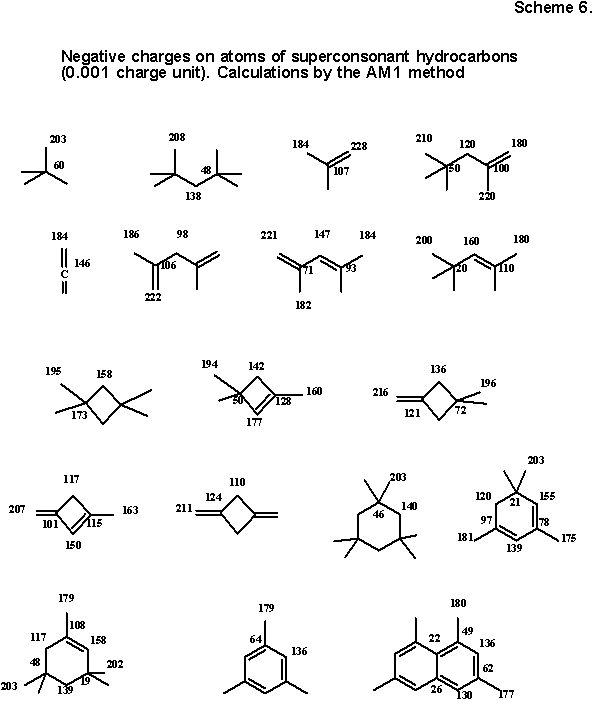
Since all carbon atoms carry a weak negative charge, it would appear that there is no alternation. In fact, alternation is displayed very specifically, namely in the clear alternation of a strong and weak charge of carbon atoms. In the overwhelming majority of cases the large minus is namely there where the hypothetical C4- anion can be localized, exceeding by approximately twice in value the weak charge of those positions, where the C4+ ion should be. (Exceptions are little expressed and pertain to nonconjugated double bonds and the nodal atom in the naphthalene nucleus.)
It is not surprising that this rule is not fulfilled for the triple bond (say, in the case of propyne). The attempt itself to relate the hidden cationoid nature with the sp-hybridization of an atom has internal contradiction: in the triple bond both atoms have the highest s-character, and consequently, a potential anionoid type. Nevertheless, a method exists of showing the hidden electrophilic character of the sp-hybridized central atom of propyne, namely without going beyond the framework of alternating charges of the semiempirical models. Charges for neutral superconsonant hydrocarbons and of their conjugated cations and anions (superconsonant by definition), obtained by deprotonation of the sp3-hybrid center, are compared in Scheme 7. It is not difficult to see that both in sigma- and in pi-systems the unexpressed alternation of the neutral hydrocarbon is transformed to a strong plus and minus gradient, observed in the ion, enveloping, in particular, also the case of the triple bond of propyne.
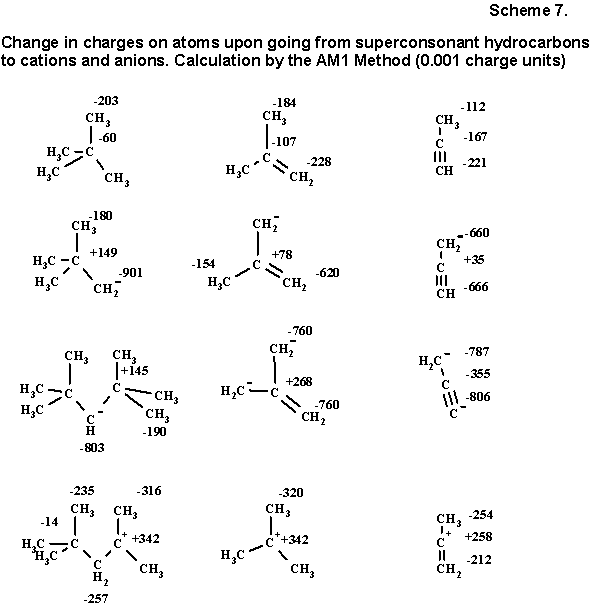
5. RETENTION OF PROPERTIES OF SUPERCONSONANCE IN POLAR PROCESSES
5.1. Preliminary observations. Continuing the analogy between superconsonant structures and ion associates, we are faced with the question: what features of chemical properties of ionic compounds can be associated globally with the chemical behavior of superconsonant structures? Evidently the main property of the ion (distinguishing it from a non-ion) is its charge. It is clear that in relation to dissociation (dissolution of a salt in water), association (formation of a crystal), or ion-exchange processes the property of the ion to carry a certain charge does not disappear. To destroy this property means to remove or add an electron, say, by electrolysis or the action of an external oxidizing or reducing agent.
This is a very trivial deduction; however, in relation to covalent superconsonant structures it has a fundamentally different shade of meaning: are strictly formal ions, collapsed into a covalent superconsonant structure, obliged to retain at least a "memory" of their charge during analogous association (addition), dissociation (elimination), and exchange (substitution) processes in the absence of external oxidizing and reducing agents? Is the topological bipartite property, assigned to such structures, where the color is rigidly associated with charge, a universal and constant invariance in the absence of redox processes? A partial answer to the question (an example that this is sometimes encountered) was given by Evans and Seebach, who noted that consonance can be created from consonance, and a normal chain can be created from another normal chain. Meanwhile, both authors did not note that they gave a constructive example of a conservation rule, namely, conservation of alternation.
In the Seebach model the normal structures (or their synthones) and certain "external" reagents (the polar structure of which is not compared with the initial reagents) exist initially. In our model the polar structure of any reactant is fundamental: it either is or is not superconsonant. The question arises: are there among the reactants, mentioned by Seebach for creation of the umpolung (see the above-enumerated 6 types) examples of superconsonant species? It is extremely evident that the use for this purpose of typically carbenoid, sextet, or septet species, cyclopropanes, and cyanide and acetylenide ions is the use of certain external (in relation to the superconsonant set itself) structures, which are not presentable as an associate of "proper" ions, but consequently act as (obvious or hidden) oxidizing and reducing agents. Redox reactions should be rejected for the same reason. Thus, only the umpolung remains by substitution of the heteroatom, which, as follows from all examples, discussed in a review [52], does not include one case of superconsonant structures. Thus, we come to the preliminary and not at all trivial conclusion that it is not possible to create an umpolung (structural dissonance, polar heterogeneity) with reactions only inside the set of superconsonant structures.
5.2. Formulation of hypotheses. We note that to date the umpolung was understood to be namely a process of creation of a structural fragment, contradicting the normal alternation in the chain. Since our definition of alternation was slightly broader from the very beginning (including the correct mutual arrangement of hydrogen atoms, and also hydrogen and heteroatoms), we were also required to analyze the umpolung more broadly, namely, as the loss of the property of superconsonance. In this connection it is lawful to ask the question: is it possible only by polar reactions and only inside the superconsonant set to emerge from its limits? If we can categorically answer negatively to this question, then this would result in the fundamental principle, which can be expressed in three equal formulations:
5.3. Features of reactivity
of superconsonant structures. We will assume the formulated principle
as a working hypothesis, with which facts can be compared. If the problem
is approached pedantically, then in more than one and a half centuries
chemists have not so frequently carried out "pure" experiments between
strictly superconsonant structures. (Traditional acids and bases, tertiary
amines, esters, many standard solvents are evidently not such structures.)
Nevertheless, as we already mentioned in Subsection 4.2, strongly consonant
structures are too well known to organic chemists, and features of their
synthesis and reactivity have long passed into classical textbooks.
The necessary facts are completely sufficient in order to be convinced of the validity of hypotheses at least for a broad class of oxygen-, nitrogen-, and fluorine-containing structures. In the majority of cases such processes are in fact typical "reactions from the textbook," discovered yet in the last century or at the beginning of this century. It is sufficient to recall, for example, the Ritter and Reformatsky reactions, individual examples of the Michael and Claisen reactions, the Favorskii rearrangement, different syntheses based on derivatives of malonic and acetoacetic acids, classical "named" ring syntheses of pyridines, pyrimidines, pyrillium salts, pyrones, etc.
It is necessary with some regret to discard positive examples of reactivity of whole classes of organic compounds (esters, ketals, vinyl ethers, derivatives of aliphatic alcohols and amines, chloro-, bromo-, silico-, boro-, and alumino-organic structures), which clearly indicate retention of consonance in polar processes, but the structure of which, however, does not fall within the framework of superconsonance.
If it is considered that the definite result of the development of organic chemistry of the first half of the century led to a classical multiple-volume series [65], then it is seen that up to 1952 in the chemistry of polar acyclic structures a worthy counterexample to the above-formulated principle simply did not exist. (At least they were not reflected in the first two volumes of the series, containing an infinite enumeration of specific reactions of superconsonant aliphatic compounds.)
We note that in the history of chemistry there is an example of the consecutive use of the principle of superconsonance in the practical design of carbo- and heterocycles. We mean the famous acetate hypothesis of Collie [66], according to which a series of aromatic and heteroatomatic structures can be made up of fragments of acetic acid. Namely this model was the basis of modern concepts in the biosynthesis of phenols [67].We will recall that namely substituted benzenes were earlier the objects of spirited discussions between supporters and opponents of the alternation model. (Thus, meta-nitration of aniline in acids or the cases of nonstandard orientations were used for rejection of the model). Meanwhile, in the case of superconsonant benzenes (e.g., 1,3,5-substituted benzenes with donor groups) the possibility of electrophilic meta-substitution in relation to the donor group is virtually absent of occurrence (this would require ipso-attack). On the other hand, the most stable arenonium ions (evidently, also superconsonant), arising at practically any combination of F, OH, CH3, and even NH2 groups, are recorded in these systems. In addition, nucleophilic substitution of such molecules, in contrast to other benzenes, does not require additional activation (e.g., cf. mutual transformations of phluoroglucinol to 1,3,5-triaminobenzene and vice versa). By the way, such structures are most stable thermodynamically and are frequently the final products during isomerization in both acidic and nucleophilic media.
Certain other famous and most vivid features of the chemical behavior of individual classes of strongly consonant systems can be given without references. Thus, the conclusion is known in the chemistry of aliphatic fluorine derivatives that fluoride ion is the "mirror" of a proton (cf. the behavior of isobutylene and perfluoroisobutylene upon the action of HF). It is not difficult to recall examples, in which the cyano group will be the "mirror" of fluorine (comp. the acidic properties of hexacyanoisobutylene or the "cyaniding" properties of tetracyanomethane in relation to fluoride ion).
A large number of reversible processes and cleavage reactions of superconsonant structures into simpler subunits with the expected retention of alternation in the chain are known (cf. the unusually easy cleavage of beta-dicarbonyl compounds, beta-keto- and beta-dicarboxylic acids, and polyacids). Pyrones and pyrillium salts undergo easy ring opening, and the benzene nucleus can be created by transformation of 2,4,6-pyrillium or collidinium salts. Somewhat exotic basic cleavage of benzene ring in phluoroglucinol into acetone, acetic acid, and CO2 is completely trivial in this context. The easy opening of superconsonant structures with four-membered ring is observed in the case of beta-lactones, dimeric isocyanates, and during Grob fragmentation of cyclobutenes.
Thus, a large number of polar superconsonant structures and polar processes exist, in which the topological property of polar dipartition and consequently, also the polar nature of centers, are retained. The majority of such reactions are ionic electro- or nucleophilic addition or substitution processes, occurring under polar conditions, including acidic or basic catalysis. Frequently these are reversible reactions, subject to charge control, to which the principle of microscopic reversibility is applicable. We have found very few exceptions (see Section 5.5).
A significant amount of data indicates that the property of superconsonance, not oddly, is also retained for nonpolar systems in polar processes. The most banal example of such a reaction is the classical experiments of Butlerov on the action of sulfuric acid on isobutylene. (It is clear that polyisobutylene or any oligomer of isobutylene, obtained by the "head to tail" type, is superconsonant). Other examples can be arbitrarily divided into three large classes.
First, these are acid- or base-catalyzed prototropic rearrangements, not changing the initial alternation, known for the whole series of purely hydrocarbon structures (acetylene-allene-diene rearrangements, migration of the triple bond in diynes and enynes, transformation of the exo-double bond to an endo-bond in 1,3-dimethylenecyclobutane, etc.).
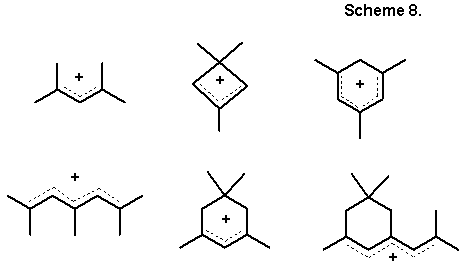
Second, these are processes of protonation and deprotonation of structures with a superconsonant arrangement of functions, exactly corresponding to the requirement of maximum delocalization of the formed charge. Thus, it is well known [68] that structures of the most stable acyclic and even-cyclic carbocations are superconsonant, Scheme 8 (comp. with calculation data in Scheme 6), while the charge can be localized namely in those positions, where the hypothetical C4+ center would be situated.
In addition, stable mono- and polycharged anions of superconsonant hydrocarbons are well known with conjugated and cross-conjugated bonds (mono- and dianion of mesitylene, dianion of isobutylene, bi- and tetra-deprotonated anions of propyne and pentadiyne, e.g., see [43]), the structure and reactivity of which completely agree with the examined principle. A curious fact of gas-phase chemistry of carbanions, generated from alkylcarboxylic acid anions [69], is the unexpected stability of namely methyl and neopentyl anions (which are superconsonant), in comparison with intermediate homologs.A large third group of reactions includes the appearance of a polar bond in the initial nonpolar substrate due to a reaction with polar reagents. Classical examples are Markovnikov addition to isobutylene and its dimers, hydration of propyne and allene, the consecutive acetylation of isobutylene to mesitylene oxide or pyrillium cation, and numerous deprotonation reactions of acetylenes (e.g., propyne) with electrophilic multiple bonds.
5.4. Retrosynthesis, synthetic equivalence, and bipartition. The broad observance of a simple rule in a series of polar molecules, the structure of which satisfies the rigid determinism of bipartite graphs, makes it possible to look anew at the problem of design of synthesis of superconsonant structures. If we limit ourselves only to "logical" (according to Corey) separation of bonds of the targeted structure, then the precursors compulsorily must be superconsonant. It is found that in the examined series the problem of retrosynthesis (to predict all possible precursors and to select the most optimal) and the problem of direct design of the result of synthesis (to predict all possible reaction products and to estimate the most probable) become symmetrical to certain degree, although by virtue of the proposed closed nature of set. At least namely in this matter is permissible the maximum convergence of positions of the abstract mathematician and the experimental chemist, the combination of empirical knowledge on the nature of things with the use of algebra and combinatorial analysis.
We will limit ourselves to the examination of one fundamental question: to what innovation can a purely mathematical model lead in the understanding of synthetic equivalence. It is well known that certain strongly consonant structures are easily transformed into one another or enter into reactions as synthetic equivalents, which is very important during the design of the synthesis of a more complex carbon chain or ring. (Thus, carbon "suboxide" replaces malonic acid or its acid chloride, acetoacetic acid is equivalent to diketene and can be reversibly transformed to the enamine, beta-halovinylcrotonic, beta,beta-dihalobutyric, or tetrolic acid.) It is not difficult to determine that the main factor, determining the equivalence of functions in the superconsonant series, is namely the identity of the carbon chain: the nature of the heteroatom and the position of the multiple bond or hydrogen atoms are frequently not fundamental. For the simplest case such equivalence can be arbitrarily expressed by Scheme 9, symbolically combining the mutually interchangeable structures into horizontal rows. In this case the rigid requirement of superconsonance of synthones can be completely removed, and the heteroatom X in structures of Scheme 9 can be replaced with certain care by the appropriate substituent (leaving group).
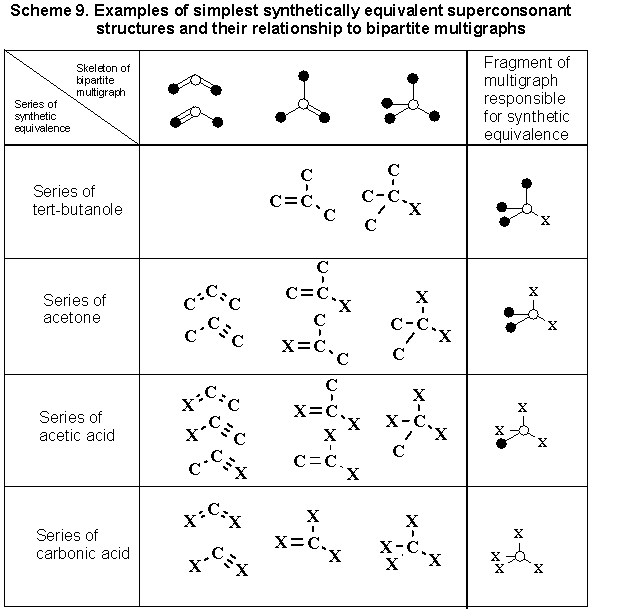
In Section 4.3 we showed the existence of a strict genetic relation between strongly consonant structures. Upon comparison of Scheme 9 with Schemes 4 and 5 it is not difficult to see that synthetically equivalent series can be described with the same ease (and generated on the computer) with the use of the same combinatorial model. It is important that in the process of such "splicing" the number of carbon atoms, responsible for the synthetic equivalence, remain unchanged. While synthones of Scheme 9 (derived from Scheme 4) are almost trivial, the synthones, which can be obtained from the multigraphs, let us say of Scheme 5 and more complex series, will already be not at all traditional. Low polarity unsaturated fragments or even rings can be unexpected equivalents of normal compounds. This problem becomes especially important during analysis of nontrivial cyclic precursors for target cyclic structures, particularly for the design of recyclizations of heterocycles [62]. Another possible application of the approach can be the use of nontrivial synthones in construction of even-ring heteroaromatic systems [58-60].
5.5. The rule and exceptions. It is natural to expect that the less polar the structure, the greater is the probability of deviation of its chemical behavior from the property to retain superconsonance (it is difficult to lose what it does not have). Analogously, disturbance of the rule can be expected in the case of nonpolar reactions. More surprising are the facts of retention of superconsonance, frequently occurring during thermolysis. In addition to "depolymerization" of di- and triisobutylene, the decomposition of 1,1,3,3-tetramethylcyclobutane [70] into two isobutylene molecules (one of the diisobutylenes is also formed on the side) can be mentioned. Decomposition of tris-(neopentyl)aluminum into trimethylaluminum and isobutylene [71] (i.e., actually "heterolysis" of neopentyl anion into superconsonant parts) also has a nonpolar analog in thermal cleavage of neopentane into methane and isobutylene.
Another factor, able to promote disturbance of polar bipartition, is the above-noted contradiction between the potential cationoid character and sp-hybridization in acetylenes and allenes. In fact, a characteristic feature of allene chemistry is thermal dimerization of the "head-to-head" type, leading to an even chain between hydrogen atoms or between heteroatoms (the reaction is typical for both allene and for perfluoroallene). We note that this fact does not exclude the possibility of making this reaction occur in the "correct" way (comp. the example of ionic dimerization of allene by the "head-to-tail" type in acidic medium with formation of superconsonant l,3-dihalo-l,3-dimethylcyclobutane [72]). The clear selectivity, observed in reactions of nucleophiles with allenes, substituted with acceptor groups, also cannot be ignored [73]. An example of the direct effect of alternation in the commulene systems is "mirror" reactions of perfluoroallene and per(trifluoromethyl)allene: the nucleophiles react with the first at the terminal atom and with the second at the central carbon atom.In the chemistry of polar reactions of alkynes, enynes, and diynes of low polarity there are unfortunately more questions than answers [73-75]. The problem is mainly the disagreement and nonpredictability of results for hydration and addition reactions of alcohols or hydrogen halides for structures of this class in general, and for superconsonant structures in the special case. For example, pentadiyne-1,3 is hydrated in acid with retention of the superconsonant motif, while alcohols of normal structure (assume that they are not superconstant) attack the terminal sp-hybrid atom [76]. For example, methylisopropenylacetylene, adding hydrogen halide (at the double bond) or alkoxide (at the triple bond) in a consonant manner, undergoes hydration at the C-3 atom with disturbance of superconsonance [75, 77]. Evidently steric factors can have a dramatic effect on the direction of reactions of acetylenes with nucleophiles. Thus, significant amounts of the anti-Markovnikov adduct appear in the reaction of propyne with the tert-butylate anion [78]. It seems that examples of new exceptions are possible in namely this region.
The weak polarity of CH-bonds of hydrocarbons under appropriate conditions can be subjected to "incorrect" heterolysis into a hydride and carbocation center upon the action of strong Lewis acids. Thus, skeletal isomerization of neopentane upon the action of aluminum halides is evidently caused by cleavage of hydride ion with formation of a dissonant neopentyl cation. Protonation of methane in superacidic media leads to generation of a dissonant methyl cation, participating in further transformations; tert-butanol in 96% sulfuric acid forms hydrocarbons for probably the same reasons.Finally, electrocyclic processes, weakly sensitive to polar factors, do not have to obey rules of regioselectivity, characteristic for ionic intermediates. An example of competition between a pericyclic reaction and stabilization of the superconsonant species is the behavior of the cation, obtained from 2,4,6-trimethylhepta-1,3,5-triene. The spectrum of the cation can be recorded, and then the structure undergoes conrotatory cyclization with closing of the dissonant 5-membered ring [68].
Thus, the discussed superconsonant set is obtained "almost closed." The chemist can retort to the mathematician that in organic chemistry two times two is "almost always" equal to four, although sometimes also to five (if nonpolar substrates are not ignored). In addition, cases of spontaneous disturbance of polar dipartition are most curious as examples of a natural umpolung, and not one introduced from outside.
5.6. Problem of new heteroatoms. Introducing the definition of strongly consonant structures, we were limited to a minimum set of atoms and ions. In principle, the model can be expanded by adding to the collection of initial ions other ions, having the closed electron shell of an inert gas. Nothing interferes with the formal description of methods of the synthesis and reactivity of such "homogeneous", but heteroatomic or organoelement systems with the use of the corresponding saturated ionic structures. The simplest example is nitromethane, in which it is easy to separate arbitrarily the N5+ ion (isoelectronic with the C4+ ion) or thiourea, "containing" sulfide ion. However, the chemistry of homogeneous compounds even only with these two ions can contain a record number of facts of spontaneous disturbance of bipartition or of the umpolung (comp. [47, 52]).
It is reasonable to separate all new ions arbitrarily into two classes. In the first class (strongly consonant ions) to the six already available ions can be added cations of alkali and alkaline-earth metals, B3+, Al3+, Si4+ (and possibly their analogs) ions, and also chloride and (with certain care) bromide ion.
The second class will include the remaining octet ions, particularly N5+, S6+, P5+ cations and S2-, B5-, I- ions, which can be arbitrarily called weakly consonant.
The fundamental difference between these classes is that consonant structures, obtained by a combination of ions of the first class, will have in the majority of cases a tendency to retain the property of polar dipartition.
The presence in a molecule of ions of the second class will lead both to retention and to partial disturbance of this rule. Without discussing in detail the reasons for this phenomenon, we note the clear parallel with the principle of HSAB and emphasize that the structure itself of ions of the second class contains an internal conflict (at least between the electronegativity of the atom, the presence of a vacant orbital, and the sign of the charge). This masked conflict in complex organic structures with covalent bonds will lead to an anomalous orientation in polar processes, the simultaneous display by the atom of electro- or nucleophilicity, i.e., to the appearance of dissonance or an umpolung.This paper was generously financed by a grant from the International Scientific Fund (ISF Grant M1X000) and a grant from the St. Petersburg Center of Fundamental Natural Science. The author is grateful to Drs. M. Saltzman (USA) and J. Sander (Germany), who kindly presented copies of little accessible publications [8, 50] and manuscripts [47].
REFERENCES1. I. Hargitai and M. Hargitai, Symmetry in the Eyes of a Chemist [Russian translation], Mir, Moscow, 1989.
2. N. F. Stepanov (Editor), Principles of Symmetry and Systematics in Chemistry, Coll. of Papers [in Russian), MGU, Moscow, 1987.
3. R. King (Editor), Chemical Applications of Graph Theory and Topology [Russian translation], Mir, Moscow, 1987.
4. M.A. Johnson and G.A. Maggiora (Editors), Concepts and Applications of Molecular Similarity, NY: J. Wiley, 1990.
5. E. V. Babaev, Khimiya i Zhizn, no. 4, p. 17, 1989.
6. R. Woodward and R. Hofmann, Retention of Orbital Symmetry [Russian translation], Mir, Moscow, 1972.
7. E. V. Babaev, Chapter 7, in: Graph Theoretical Approach to Chemical Reactivity (Understanding Chemical Reactivity, Vol.9), D. Bonchev and O. Mekenyan (Editors), Dordrecht-Boston-London: Kluwer Academic Publ., p. 209, 1994.
8. A. Lapworth, Manchester Memoirs Lit. Phil. Soc., vol. 64, no. 3, p. 1, 1920.
9. W. O. Kermack and R. Robinson, J. Chem. Soc., p. 427, 1922.
10. C. K. Ingold, J. Chem. Soc., p. 513, 1925; Ibid., p. 870, 1870.
11. A. Lapworth, Chem. Ind. (London), p. 563, 1925.
12. Chem. Ind. (London), p. 1050, 1925.
13. J. Shorter, Nat. Prod. Reports, vol. 4, p. 61, 1987.
14. C. A. Russel, Ibid., p. 47; M. D. Saltzman, Ibid., p. 53; W. Cocker, Ibid., p. 68.
15. M. Saltzman, J. Chem. Educ., vol. 49, no. 11, p. 750, 1972.
16. C. B. Flurcheim, J. Pract. Chem., vol. 76, p. 185, 1907.
17. D. Vorlander, Ber., vol. 52, p. 263, 1919.
18. J. Steiglitz, J. Amer. Chem. Soc., vol. 44, p. 1293, 1922.
19. K. Fajans, Chem. Eng. News, vol. 27, no. 13, p. 900, 1949.
20. W. Huckel, Theoretical Fundamentals of Organic Chemistry [Russian translation], Izdatinlit, Moscow, vol. 2, pp. 523, 535, 1958.
21. E. Muller, Neuere Anshanungen der Organischen Chemie, Berlin: Springer, p. 66, 1940.
22. A. E. Remick, Electronic Interpretation of Organic Chemistry, NY: J. Wiley, p. 27, 1947.
23. J. B. Conant and W. R. Kirner, J. Amer. Chem. Soc., vol. 46, p. 232, 1924.
24. J. B. Conant and R. E. Hussey, J. Amer. Chem. Soc., vol. 47, p. 488, 1925.
25. E.L. Eliel, in: Steric Effects in Organic Chemistry, M. S. Newman (Editor), NY: J, Wiley, p. 138, 1956.
26. A. Iliceto, A. Fava, and A. Simeone, Gazz. Chim. Ital., vol. 90, no. 4, p. 660, 1960.
27. K. L. Williamson, S. Mosser, and D. E. Stedman, J. Amer. Soc., vol. 93, p. 7208, 1971
28. I. Morishima, T. Yoshikawa, et al., J. Amer. Chem. Soc., vol. 95, p. 165, 1973.
29. D. W. Davis, M. S. Banna, and D. A. Shirley, J. Chem. Phys., vol. 60, p. 237, 1974.
30. E. E. Ernstbruaner and J. Hudec, J. Amer. Chem. Soc., vol. 96, p. 7106, 1974.
31. J. E. Lambert, D. A. Netzel, H. Sun, and K. K. Lilianstrom, J. Amer. Chem. Soc., vol. 98, p. 3778, 1976.
32. L. P. Zakulaev, R. P. Vorob'eva, Zh. V. Shmyreva, and T. A. Oleinikova, Zh. Obshch. Khim., vol. 44, no. 5, p. 1141, 1974.
33. S. Oae and C. A. van der Werf, J. Amer. Chem. Soc., vol. 75, p. 5037, 1953.
34. R. D. Stolov, P. W. Samal, and T. W. Giants, J. Amer. Chem. Soc., vol. 103, p. 197, 1981.
35. I. A. Leenson, Even or Odd? [in Russian], Khimiya, Moscow, 1988.
36. A. A. Petrov, Chemistry of Alkanes [in Russian), Nauka, Moscow, 1974.
37. M. Dewar and R. Doherty, Theory of Perturbation of Molecular Orbitals in Organic Chemistry [Russian translation], Mir, Moscow, 1977.
38. J. A. Pople and M. S. Gordon, J. Amer. Chem. Soc., vol. 89, p. 4253, 1967.
39. W. J. Hehre and J. A. Pople, J. Amer. Chem. Soc., voL 92, p. 2191, 1970.
40. P. Politzer, S. L. Whittenburg, and T. Warnheim, J. Phys. Chem., vol. 86, p. 2609, 1982.
4I. G. van Hooydonk, J. Mol. Struct. (THEOCHEM), vol. 121, p. 45, 1985.
42. S. W. Benson, Angew. Chem. Int. Ed. Engl., vol. 17, p. 812, 1978.
43. J. Klein, Tetrahedron, vol. 44, no. 2, p. 503, 1988.
44. T.-L. Ho, Reviews of Chem. Intermediates, vol. 9, p. 117, 1988.
45. T.-L. Ho, Polarity Control for Synthesis. Chichester: J. Wiley, 1991.
46. E. J. Corey, Pure Appl. Chem., vol. 14, p. 19, 1967.
47. D. A. Evans, Manuscript 242A: D. A. Evans, An Organizational Format for the Classification of Functional Groups. Application to the Construction of Difunctional Relationships, UCLA Physical Organic Chemistry Seminar, May 6, 1971.
48. L. Ugi and P. Gillespie, Angew. Chem., vol. 83, no. 23, p. 985, 1971.
49. R. Doenges, B.-T. Groebel, H. Nickelsen, and J. Sander, J. Chem. Inf. Comput. Sci., Vol. 25, no. 4, p. 425, 1985.
50. F. Serratosa, Buttleti Societat Catalana Cienc. Fis., Quim., Mat., vol. l, no. 2, p. 75 (Chem. Abstr. 89: 196341 s.), 1977.
51. D.A. Evans and G. C. Andrews, Acc. Chem. Res., vol. 7, p. 147, 1974.
52. D. Seebach, Angew. Chem. Int. Ed. Engl., vol. 18, no. 4, p. 239, 1979.
53. M. Wagener and J. Gasteiger, in: Software Developments in Chemistry, J. Gasteiger (Editor), Springer-Verlag, p. 265, 1990.
54. F. Serratosa, Organic Chemistry in Action. The Design of Organic Synthesis, Amsterdam: Elsevier, 1990.
55. A. McKillop and A. J. Boulton, in: Comprehensive Heterocyclic Chemistry, A. R. Katritzky and C. W. Rees (Editors), Oxford: Pergamon Press, vol. 2, p. 67, 1984.
56. E. V. Babaev and S. V. Tsitovskii, Vestnik MGU. Seriya 2 (Khimiya), vol. 35, no. 1, p. 85, 1994.
57. E. V. Babaev, 1st European Conference on Computational Chemistry, Nancy. Abstr., p. 26, 23-27 May 1994.
58. E. V. Babaev, KhGS, no. 7, p. 937, 1993.
59. D. E. Lushnikov and E. V. Babaev, KhGS, no. 10, p. 1299, 1993.
60. E. V. Babaev, KhGS, no. 7, p. 962, 1993.
61. F. Harari, Graph Theory [Russian translation], Mir, Moscow, 1973.
62. E. V. Babaev, D. E. Lushnikov, and N. S. Zefirov, J. Amer. Chem. Soc., vol. 115, p. 2416, 1993.
63. E. V. Babaev, in: Collection: History and Methodology of Natural Sciences, no. 35, Philosophic Problems of Chemistry [in Russian], A. P. Rudenko (Editor), MGU, Moscow, p. 1221, 1988.
64. K B. Wiberg and P. R. Rablen, J. Comput. Chem., vol. 14, no. 12, p. 1504, 1993.
65. E. H. Rodd (Editor), Chemistry of Carbon Compounds, Amsterdam: Elsevier, vol. la, pp. 221-777, 1951; vol.lb, pp. 779-1462, 1952.
66. J. N. Collie, J. Chem. Soc., vol. 91, p. 1806, 1907.
67. A. J. Birch, Science, vol. 156, p. 202, 1967.
68. Modern Problems of Physical Organic Chemistry [Russian translation], Mir, Moscow, p. 393, 1967.
69. R. R. Squires, Acc. Chem. Res., vol. 25, p. 461, 1992.
70. T. A. Babcock, J. Amer. Chem. Soc., vol. 91, p. 7622, 1969.
71 K. Ziegler, Angew. Chem., vol. 68, p. 721, 1956.
72. K. J. Griesbaum, Amer. Soc., vol. 86, p. 2301, 1964.
73. Houben-Weyl, Methoden der Organischen Chemie. Stuttgart, Thieme, vol. 5/2a, 1977.
74. The Chemistry of Carbon-Carbon Triple Bond, (The Chemistry of Functional Groups), S. Patai and (Editor), Chichester, J. Wiley, parts 1, 2, 1978.
75. A. A. Petrov, Usp. Khim., vol. 29, no. 9, p. 1049, 1960.
76. B. P. Gusev and V. F. Kucherov, Izv. AN SSSR. Ser. Khim., no. 1, p. 208, 1974.
77. B. S. Kupin and A. A. Petrov, ZhVKhO im. D. I. Mendeleeva, vol. 29, p. 3738, 1959.
78. V. I. Laba, A. A. Kron, S. P. Sitnikov, and E. N. Prilezhaeva, Izv. AN SSSR. Ser. Khim., no. 9, p. 2129, 1972.